Introduction
Scope and Specificity
Among the neurodegenerative diseases that occur in mammals, the prion diseases are unique for being transmissible, for manifesting in diverse phenotypes from just one etiologic factor, and for the pathological phenomenon of “spongiform” damage to the affected tissue. In humans, the transmission also occurs genetically and spontaneous outbreaks seem to occur about every seven generations or so.
In animals, prion diseases are both naturally-occurring and thought to be transmitted by ingestion of feed with animal byproducts. Examples of the former are chronic wasting disease (CWD) endemic to North American deer and elk, as well as “scrapie” in sheep and goats. The transmissible variants include feline spongiform encephalopathy (FSE), bovine spongiform encephalopathy (BSE), and transmissible mink encephalopathy (TME).
As to humans, the spectrum of prion disease comprises iatrogenic CJD (iCJD), kuru, Gerstmann-Sträussler-Scheinker (GSS) disease, sporadic Creutzfeldt-Jakob disease (sCJD), familial CJD (fCJD), fatal insomnia (FI), and new variant CJD (nvCJD or vCJD).
The Nature and Structure of Prions
Prions are infectious agents with an extracellular form that may or, more likely, does not contain nucleic acids. Even if prions do possess trace nucleic acids, it is speculated, these are not long enough to assemble into a strand that can encode the protein it is with. Not having nucleic acids of their own, prions propagate within the genomic material of living organisms (Qin, 2006).
Following an investigation into the infectious agent responsible for scrapie in sheep, Prusiner (1982) conceived “prion” for “protein-like infectious agent.” Even earlier, work on determining the nature of this curiously atypical pathogen had already revealed that a structure with nucleic acid could be ruled out since scrapie fractions remained virulent even when exposed to ultraviolet or ionizing radiation (Alper, Cramp, Haig, and Clarke, 1967). Subsequently, (Prusiner, 1982; McKinley, Bolton, and Prusiner, 1983), eminently satisfactory results with reducing infectivity were attained when fractions were exposed to high heat, chemicals such as formalin or diluted for prolonged periods in proteases. While these strongly suggested a protein structure, Prusiner et al. (1981) discovered just how robust this pathogen could be on consistently finding a protease-resistant protein in the brains of humans and animals that had succumbed to Transmissible Spongiform Encephalopathy (TSE).
Prions are a substructure of viruses and are distinguished from viroids in that the latter is formed of RNA in circular structures while prions are unique for a structure that is wholly “misfolded” protein and that exists in extracellular space.
Epidemiology
The Single Etiology of Prion Diseases
Prior to the work of Prusiner in learning the probable composition of prions, little was known beyond the decades-old speculation of Sigurdsson (1954) that TSE’s seemed to have in common a slow-acting virus that was transmissible but had a long incubation period. Taking his cue from Alper et al. (1967) that the nucleic acid that would enable reproduction might be missing, Prusiner generated findings that leaned towards the pure protein theory. Regardless, Mastrianni and Roos (2000) contend, the composition debate is by no means settled.
The Mutation of Normal Expressed Protein to Transmissible Pathogen
Granting a pure-protein structure, transmissibility is therefore reliant on the mutation of the protein-producing PRNP gene. A great deal of information has already emerged in the literature regarding the structure of the prion protein (PrP). It has been found that the misfolded isoform (PRNP) of the naturally occurring PrP can trigger normal PrP to misfold in the same way. Over time, the in-vivo amplification of PRNP destroys large numbers of neurons. Eventually, the brain tissue becomes sponge-like, with vacuoles and plaques distributed throughout all regions of the brain (see also section D., “Stages of Prion Infection” below). The PRNP isoform is also found in peripherally circulating white cells and platelets.
Once some consensus had formed about the protein stricture of prion, it was necessary to establish that the transmissible pathogen (originally associated with scrapie, hence the designation PrPSc) is merely an isoform of normally-expressed, cellular PrP or PrPC. Both isoforms boast identical amino acid sequences but could not be more different biochemically. PrPC dissolves in nondenaturing detergents and in the presence of proteases; in contrast, PrPSc is insoluble to the first and comparatively resistant to the second (Mastrianni and Roos, 2000).
Structural studies by Safar et al. (1993) also reveal that normally-expressed PrPC takes a largely-helical shape, while pathogenic PrPSc resembles pleated sheets in form. It appears that this structural alteration is the signal event in the progression of prion disease because little is known about what brings about the destruction of neurons.
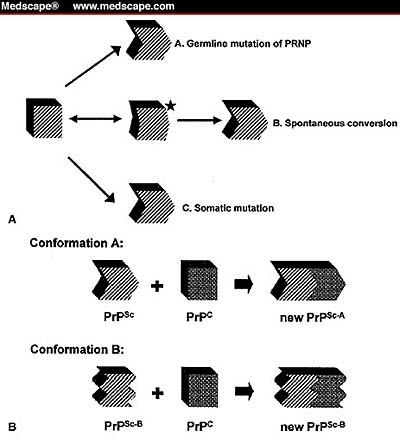
Similarly, the mechanisms that trigger the conversion of PrPC to PrPSc constitute a gray area. As illustrated in Figure 1 below, there are four hypothesized mechanisms. First is the mutation of the human prion protein gene (PRNP) germline mutation. This destabilizes PrPC and paves the way for PrPSc production. Mastrianni and Roos suggest that this is the mechanism at work in genetic prion disease (2000).
The second possibility is a somatic mutation that may trigger the pathogenic conformation of PrP or occur as an idiosyncratic metamorphosis of PrPC that does not refold as cellular PrP would.
However mutation is triggered, the solitary infectious PrPSc interacts with surrounding PrPC as a kind of template for transforming more PrPC to the pathogenic isoform. Such induced mutation explains why it is not necessary for the infectious agent to be self-replicating. At the same time, the progressive loss of surrounding neurons brings about the characteristic appearance of vacuoles in the brain tissue of TSE patients.
In the middle path below, thirdly, Mastrianni and Roos (2000) conjecture that for some reason as yet unknown, PrP may transition to an unstable form that mutates to either the cellular or pathogenic isoforms. This presumes undiscovered factors that trigger a destabilized PrP.
Modes of Transmission
The highly contagious nature of Prion diseases, transmissibility by ingestion, and ability to make the cross-species “jump” have aroused great concern among epidemiologists and the public at large. For instance, “Bovine Spongiform Encephalopathy” (BSE) periodically re-surfaces, runs rampant in cattle, and generates intense attention in media as “mad cow disease.” In humans, the BSE-originating prion manifests as “variant Creutzfeldt-Jakob disease” (vCJD) because it is literally a variant of CJD. There is still a considerable debate and speculation around sparse scientific evidence on the matter of prion disease transmissibility from animals to humans. Beyond BSE, a review by Will (1993) has classed scrapie in sheep, Transmissible Mink Encephalopathy (TME), Chronic Wasting Disease (CWD) in elk and deer and Feline Spongiform Encephalopathy in cats as likely transmissible from animals to man.
That prion disease is transmissible at all had already been established as far back as 1939 when Cuillé and Chelle induced scrapie in goats by intraocular injection of scrapie-infected spinal extract. The prevalence of kuru among the Indigenous tribes of New Guinea was traced to cannibalism and specifically the ingestion of non-muscular tissues of already infected victims. This was the cue for Gajdusek, Gibbs and Alpers (1966) to successfully test kuru as pathogenic in both humans and animals, in this case, chimpanzees.
Blood Borne Transmission of CJD
In 2004, Peden and Lleweylln separately published their findings describing the possible transmission of vCJD via blood transfusion. Both researchers described persons in whom vCJD onset was less than 10 years after the receipt of packed red blood cells (which were later found to be positive for CJD). In both investigations, both donor and recipient died of vCJD confirmed postmortem.
Later studies expanded on the above to reveal that vCJD could be transmitted via straightforward blood transfusions. The ease with which the vCJD is detected in lymphoid tissue and circulating lymphocytes and the existence of a prionemia as the agent travels from the original site of the lumen of the gut to the brain brought the blood transmissibility aspect to the forefront with researchers and regulatory agencies alike. In 1999, blood donor screening precautions were implemented by the U.S. Food and Drug Administration (FDA) to exclude donors who had traveled to the UK or other European countries.
Given this route of transmission and the possibility of sub-clinical infection in human reservoirs, it has been suggested that nucleic acid testing (NAT) become mandatory in blood donor screening. Until recently, Hewitt reports, no decision had been made notwithstanding the clear risks to transfusion medicine personnel and to future blood product recipients (Hewitt 2006).
Re-Emergence
The factors which have contributed to the re-emergence of prion diseases are varied and specific to the particular types of prion disease. Yet many are conditions closely related to modern life that have contributed to the re-emergence of both human and animal prion diseases.
As mentioned in the description of prion disease, Scrapie, a prion disease of sheep, was first described in the 1800s. Scrapie is a naturally occurring disease of sheep and goats that causes ataxia, behavioral changes, and severe pruritus that leads to scraping behavior, from which the disease was named. In the 1950’s feed manufacturers in European countries, in order to become more profitable, making it a common practice to include sheep carcasses in cattle feed as a source of protein and other nutrients. Because prion diseases are transmissible via food-chain infection, the cows that were fed prion-infected sheep carcasses were at risk of developing bovine spongiform encephalopathy (BSE), the bovine variant of prion disease.
It was in 1985 that the first cases of BSE surfaced in the UK, over the next 10 years, a million cows demonstrated symptoms of BSE. There has been no evidence of horizontal transmission of BSE among cattle, and as of writing there has been no evidence of vertical transmission as well, this means that food-chain-related transmission is the single mode of infection in cows.
From Sheep to Cattle
In 1988, the practice of adding sheep offal to cattle feed was banned in the UK. This did not immediately have the desired effect since it was already known even then that there was a 4-6 year incubation period before neurological symptoms of prion disease presented. In 1992, there were an estimated 36,000 BSE-infected cows in the UK; prevalence continued to rise and by 2001, reached more than 50% of all dairy herds. The entire infected population had to be destroyed. Cases of BSE then declined at a rate of about 10% per year after that (Collee, 1997).
But another threatening omen of human infection surfaced in cats that had previously been fed food containing slaughtered beef offal. This lead to the hypothesis that other species such as humans may become infected by ingesting prion-infected beef (Bren, 2001).
After the re-emergence of BSE, human cases of prion disease recurred in the UK, Germany, Ireland, Japan and the United States. Epidemiological and laboratory studies succeeded in correlating human prion disease and the consumption of cattle that were infected by BSE. In addition to demonstrating a food chain method of transmission, a human prion disease was found to be transmissible vertically, from mother to child and horizontally through exposure to blood products. Because of the extended incubation period, infected individuals would unwittingly infect others by donating blood or blood products, such as human growth hormone, prior to becoming symptomatic (Hewitt, 2006). Furthermore, mothers of young children who presented with the neurological symptoms of prion disease shortly after giving birth unwittingly passed the disease to their offspring via breast milk (Masters 1991).
These events indicate that the global trade in medicinal products and technological advances in medicine (i.e. transplants and induction of blood products) are factors in the worldwide spread of animal and human prion diseases.
Prion/Host interactions
Insights into the pattern of human prion disease re-emergence However, to assess prion disease as a re-emerging human disease, it is helpful to understand the interactions between the prion proteins and their hosts. The latency period of the transmissible spongiform encephalopathies can be ascribed to the slow propagation of the prion protein through the peripheral and central nervous system tissues. The prion must be dispersed through an intercellular mechanism employing pathogenic, conformational, and irregular folding of the cellular prion protein. The long incubation period of 4 to 50 years can be attributed to the length of time needed for cellular display of neurological plaques and can be directly related to the dose received and point of entry (Wickner, 2004).
The case of kuru re-emergence in Papua New Guinea, some 30-50 years after the practice of consuming the entrails of deceased family members stopped, allows researchers to track this prion disease more accurately. For example, the factors that come into play with the re-emergence of kuru are strictly genetic in nature, creating the suspicion that additional cases of familial prion disease will again manifest around the globe (Collinge, 2006).
A number of contributing factors have been implicated in the re-emergence of modern-day prion disease. These factors include genetics, human/animal interaction, globalization (specifically liberalized trade in food, medical supplies), increased international travel, technological advances such as transplants and global distribution of blood products, culture, and governmental intervention and regulation.
A large number of these influential variables and the simple fact of latent expression of clinical disease mandates that communication channels and appropriate infrastructure for close surveillance must be developed to support efforts to address the identification of re-emerging prion diseases.
Stages of Prion Infection
The prion pathogen triggers degenerative tissue damage that has four-fold characteristics:
Spongiform Change
The general designation of prion diseases as “transmissible spongiform encephalopathies” (TSEs) derives from postmortem findings of the universal effect on the brain. When a prion defeats the enzyme defenses of a cell, it breaks the nerve cell into fragments that fill the cell and leave holes, hence giving the surrounding gray matter or neuronal tissue a sponge-like appearance.
Even under a light microscope, it is obvious that the gray matter of the brain has suffered vacuolation. These lacuna-like gaps are illustrated in Figure 2 below. The matter of transmissibility has already been established (sections B and C above).
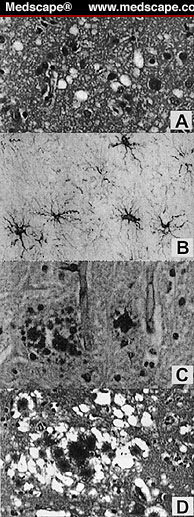
Examples of spongiform vacuolation revealed in the figure alongside are for: A) the cerebral cortex of a patient who had succumbed to sCJD shows spongiform vacuolation but without evident plaques; B) Thalamus tissue demonstrating hypertrophy and spread of astrocytes, as well as neuronal loss; C) Cerebellum tissue stained with PAS shows multicentric GSS plaques; D) In vCJD, one sees extravagant flourishing of plaques, cores of amyloid deposits inside the characteristic vacuoles.
Neuronal loss
Lacking the ability for mitosis, destroyed neurons are irreplaceable. Hence, there is a progressive degeneration of nerve function.
Astrocytosis
The astrocytes that infrequently turn into neurons proliferate, precisely because the disease has destroyed the neurons.
Amyloid plaque formation
Entangled amyloid protein turns to plaque in the aftermath of prion attack.
These four-fold characteristics of prion diseases are consistent for all species afflicted. Such neuropathological features form the basis of the histological diagnosis of human prion disease for many years, but it was accepted that these changes vary widely from case to case and even within the central nervous system in individual cases. Clinical diagnosis on gross examination remains a challenge, however.
Prevalence and Routes of Infection
At last count, Brown et al. (1987) reported that prion diseases have been recorded in virtually all countries around the world. Incidence was about the same then for both the sporadic and familial types: roughly 1 per 107 persons annually.
Across patient socio-demographics, the absence of any gender skew is counterbalanced by age bias. The inherited class of prion diseases strike at a younger age than do sporadic variants which are apt to present in patients in their 70s.
Other than iatrogenic exposure, Mastrianni and Roos report that the sole known environmental pathway for transmission is BSE exposure, which presents in humans as vCJD (2000).
Populations at Risk of Prion Disease
There are three, discreet prion disease-related, human health risks connected with environmental transmission. First, there is the iatrogenic transmission of CJD. Second, vCJD occurs from exposure to bovine by-products contaminated with BSE. Thirdly, there is a hypothesized (though not yet rigorously validated) transmission of Chronic Wasting Disease (CWD) to humans from free-ranging elk and deer (Belay, 2004).
Occurrences of iatrogenic transmission via Human Growth Hormone replacement and tissue transplantation appear to have declined at present due to increased regulatory controls put in place since 1997. However, given the long-term incubation period associated with prion diseases, many more cases can be anticipated in the future.
Risk to Blood Transfusion Recipients
As a result of the large number of human blood transfusions which occur annually, the iatrogenic blood-borne transmission of vCJD is projected to present a greater risk to populations worldwide (Aquzzi, 2006). Based on the 2004 findings of a study conducted by the Transfusion Medicine Epidemiology Review (TMER) or “Look-Back”, three cases of transfusion-associated vCJD have already been found. All three were transfused with non-leucodepleted or non-leucoreduced, red cell products. The key to this vigilance is that the probability rate of transmission via blood transfusion is 11.54% rate of infectivity. This means that 3 in 26 individuals transfused with infected blood will receive the infection. The predicted subsequent infections as demonstrated by transfusion models with sheep is 50% infectivity at year 10 (Goodnough 2004; Llewellyn 2004; MacGregor 2004).
In addition to the direct findings of TMER, it has long been hypothesized that transmission by blood transfusion could be validated by specific features of prion disease itself. CJD is predictably detected in lymphoid tissues, leading to the conclusion that circulating lymphocytes would likely be found for prion disease. It would also be logical that circulating lymphocytes would transport infected cells from the blood to the brain. Such apprehensions led to a 1999 recommendation by the Transmissible Spongiform Encephalopathy Advisory Committee and the promulgation of Federal Drug Administration (FDA) policy that blood donors who have previously spent specific periods of time in the UK and other European countries be deferred from donating blood and blood products (Federal Register, 1999).
Due to the extensive incubation period of prion diseases and the large number of transfusions that occur worldwide, additional measures have been implemented to restrict donors who may have been previously exposed to the CJD. Among those who are at greater risk are residents of the UK with higher rates of potential exposure to BSE-infected beef products (Prowes, 2002). In the UK, plasma for fractionation is no longer obtained from local residents. Since October 1999 fractionated plasma products have been imported from countries with demonstrably lower incidence of BSE and therefore potentially decreased exposure to vCJD. Blood donors in the UK are also deferred from donating blood when they have themselves been transfused after 1 January 1980 or have been recipients of allogenic tissue grafts (Prowse, 2006).
Prevention
Prions are highly resistant to in vivo and in vitro inactivation. In vitro inactivation is limited to removal rather than inactivation. It was believed early on that the leuco-reduced blood products would remove the greatest number of infectious particles. Yet studies show that this method is ineffective in that it most likely only reduces approximately 50% of the infectivity present (Grefori, 2004). Improved filters for leucocytes are being commercially developed but at present, there is still a need to filter platelets and red cells. Unfortunately, no such filter exists.
Aside from screening donors for “high-risk” factors, commercial tests are being developed to screen blood and blood products prior to transfusion. But low numbers of infectious prions usually present in the circulatory system create issues of assay sensitivity at the same time that polymorphic-type variations create problems of specificity in creating accurate laboratory screening and diagnostic assays (Brown et al., 2003).
Yet, it has been suggested that in the absence of laboratory screening assays, the most effective means by which high-risk populations can be ensured of decreased exposure is to require that blood transfusions are only given when medical necessity has been absolutely demonstrated. Regulatory agencies also require annual reporting of red cell usage and the development of programs for “better blood transfusions”. Also regulated is the use of pooled apheresis products and any effort to minimize the plasma content of platelet and red blood cell products.
A final consideration is that assessing the success of policy and institutional preventive measures is a lengthy process, given the lengthy incubation periods of prion disease and the lack of clear data regarding the sub-clinical disease.
General Effect of Prion diseases
Besides causing serious degenerative illness to the host and, at worst, fatalities over a short course of from four to six months, the prion organism is capable of mass replication. Prion proteins must be encoded with nucleic acid, and this is supported by the fact that prion-infected cells contain genes in one chromosome that encodes proteins very similar to prion protein. Apparently, prion proteins change host cell protein during or after synthesis by a series of folding actions which render them insoluble and resistant to proteases. This is the main difference between prions and viroids. The latter infects the host cell and the end product is mass production with cell lysis while the prions cause a normal host gene to create more copies of the protein itself without necessarily destroying the cell.
Those infected by TSE suffer from personality changes, psychiatric problems like depression, lack of coordination, and ataxia. Patients tend to be confused, suffer acute insomnia and myoclonus (involuntary twitching in a muscle or muscle group). In the later stages of the disease, patients have a severe mental impairment (dementia) and lose the ability to move or speak (Collinge, 2001).
Diagnostic and Surveillance Methods
Context and Background
Owing to the sheer cost associated with neurological and neuropathological examinations and diagnostics of Transmissible Spongiform Encephalopathies (TSE), only the highly industrialized and wealthy countries have the resources to participate in surveillance programs for prion diseases. Since the discovery of the new variant CJD (vCJD) in 1996, surveillance has had to become a global endeavor, largely supported by the World Health Organization (WHO). As a result, both EuroCJD and NeuroCJD have become fully operational as surveillance networks. Included in these two surveillance groups are Australia, Austria, Canada, France, Germany, Italy, the Netherlands, Slovakia, Spain, the UK, Denmark, Finland, Greece, Iceland, Ireland, Israel, Norway, Portugal and Sweden (Brown et al., 2003).
In addition, there are emergent projects such as “Prion-Net” and “SRR-CJD”, both funded by the European Commission (EC) but conducted in collaboration with WHO. These organizations study the neuropathology of human TSE and perform surveillance operations for CJD in China, Central Europe and Eastern Europe. The United States and Argentina also support and operate surveillance systems in conjunction with the aforementioned groups.
Surveillance and Diagnostic Methods
As a rule, methods used in surveillance of TSEs are highly dependent on laboratory analysis and clinical presentation. This makes observations and reporting by clinicians and pathologists of vital importance. Clinical symptoms such as dysphoria, balance and gait disturbances, memory loss, delusions, disorientation, hallucinations, involuntary movements and urinary incontinence are often presenting symptoms that arouse suspicions of vCJD and other prion diseases. At which time, neurologists and other specialists are called in to consult and rule out such confounding diagnoses such as psychiatric illness, dementia, Alzheimer disease, Wilson’s disease, vitamin B12 deficiency and/or cerebral-vascular accident, using tests such as Cerebral Spinal Fluid (CSF) analysis, Magnetic Resonance Imaging (MRI) and an electroencephalogram (EEG) to distinguish TSE from other biological and chemical processes (Collinge, 2006; Brown et al., 2003).
Surveillance System Design and Strategies
The core requirements in the design of surveillance programs for human prion disease are
- use of a common case definition;
- internationally-agreed surveillance methods;
- pooled resources for developed expertise (i.e. common laboratory and clinical reference center);
- common and accurate information reporting;
- common post-mortem service; and
- required reporting by law (Brown et al., 2003).
There are three strategies used for surveillance of human prion diseases:
- Passive – referrals are received by a central site and reports are confirmed;
- Active: reports are proactively sought from clinicians who most likely would be the first person to see the initial presentation of a case. Advertising through direct mail, internet, and professional organization participation. In addition, customized methods directed toward specific, at-risk populations are employed, particularly within ongoing cohort studies. Families of previously identified victims, recipients of blood and tissue from infected individuals, persons living proximally to zoonotic outbreaks (i.e. regarding diseases transmitted from animals to humans such as the recent BSE outbreak in England), and healthcare personnel who may have been inadvertently exposed, are all populations to be targeted and who are defined as “at-risk”. (Bernolli 1977; Aquzzi 2006; DeHayen 2006).
- As well, epidemiologists commonly resort to retrospective review of death certificates when attempting to identify epidemiological aspects of emerging events. Such retrospective reviews are of value when trying to establish timelines, infectivity rates, incubation times and sentinel cases. However, there can be legal drawbacks in investigations using death certificates. There are also countries and governments where death certificates may be unreliable as sources of documentation (Brown et al., 2003).
The Catchment Basin Approach
If resources are available, the ideal circumstance for surveillance would be to observe and monitor entire national populations. Since such an ambitious effort may not always be feasible, epidemiologists compromise for a population segment, region, healthcare system, ethnic group or other at-risk population. The choice is then defined as the catchment basin of the surveillance net.
In the case of prion diseases, population segment surveillance may include testing of blood products, tissue, bone marrow and solid organs for transplant and prior to providing the recipient with the biological product. Recently, tests have been developed that may be useful in screening donors or donor products in a cost-effective manner, much like current screening programs for hepatitis and other blood-borne infectious diseases (Cai, 2005; Warwick, 2005; Alder, 2007).
In addition, monitoring of animals such as cattle and sheep is an important point in the epidemiological surveillance process. Having oversight for the safety of food and food products, the Food and Drug Administration (FDA) and similar agencies are continuously testing the animal populations prior to distribution for food and breeding (Konold, 2006).
Screening Tests for Prion Diseases
Routine blood tests such as hematology, biochemical and inflammatory marker assays are traditionally employed in many healthcare settings. These assays are of limited use In the effort to screen for human prion disease. For instance, liver function test results are mildly elevated in prion disease. Yet, elevated liver function assays are more commonly associated with a number of other diseases such as hepatitis, HIV, cancers and drug-induced states (Alder 2007).
Additionally, assays designed to detect and diagnose diseases of the cerebrospinal fluid (CSF) are also of little use. In CJD, for instance, there are usually no indicators of inflammatory disease. Nor is there evidence that the detection of oligoclonal bands in the CSF is necessarily significant in screening for cases of CJD. Some authors suggest secondary protein studies, such as isolating the 14-3-3 protein which is elevated in some CJD patients. Yet, Herpes Simplex, stroke, subarachnoid hemorrhage, hypoxic brain damage and a number of other CSF diseases also demonstrate increased levels of 14-3-3 protein (Mastrianni and Roos, 2003)
There are clinical tests that may be helpful in the differentiation and diagnosis of prion disease, though these are not broadly applicable for screening large, asymptomatic populations. These assays are MRI, EEG and immunohistochemistry (IHC) on brain tissue biopsy (Brown et al., 2003).
Nonetheless, screening tests for donor blood, tissue and marrow bear thinking about for specificity and potential for mass deployment. These are laboratory tests utilizing DNA and protein amplification methods, such as polymerase chain reaction (PCR), that have proven to be a useful diagnostic tool in cases of prion disease and will most likely be successfully utilized in large-scale screening of donor blood and tissue. The sensitivity and specificity of these assays are currently adequate for screening purposes though one must concede that commercially developed assays are still under investigation by the FDA (Barletta 2006).
Genetic Screening
Single-amino-acid deletions and substitutions (i.e. point mutations) in the PrP have been shown to suggest susceptibility or resistance to neurodegenerative disease caused by prion amplification. In German and Icelandic populations, the genetics of human prion disease has been studied and found to be useful in identifying “at-risk” individuals who inherited homozygosity for methionine at the common polymorphism in codon 129. Heterozygotes for this polymorphism are assumed to be at reduced risk for clinically evident prion disease.
In addition, Icelandic populations were found to have a different polymorphism of PrP that appears to suggest some protective heritability against prion disease progression. This isoform of PrP was observed to be present in 46.6% of the randomly-chosen Icelandic population, where little if no evidence of prion disease has been found. Of course, further studies may be necessary but future screening tests for prion disease susceptibility may be on the horizon (Concepcion, 2002; Vollmert 2006; Georgsson, 2006).
Phenotypes of Prion Disease
This paper groups the prion diseases according to three major phenotypes by route of transmission: sporadic, familial and acquired. One concedes that the sheer variety of diseases and differences in the presentation can make rigorous classification by clinical phenotype a complex task.
Other researchers (e.g. Mastrianni and Roos, 2000) have opted for pathological profile phenotypes. On this basis, the major divisions are represented by CJD, fatal insomnia (FI), kuru, vCJD and GSS. In CJD, there is widespread gray matter spongiosis and PrP-amyloid plaques are rare. On the other hand, GSS exhibits marked PrP-amyloid deposition but minimal damage that leads to the characteristic spongiform appearance. In addition, at least three sub-variants of GSS share with Alzheimer’s disease the tangles of neurofibrillary tissue characteristic of the latter (Giaccone et al., 1990; Hsiao et al., 1990).
Unique to the Fore tribesmen of New Guinea, kuru was the first human TSE identified. This phenotype exhibits three of the pathological characteristics of TSE: marked spongiform appearance, neuronal dropout, and deposits of dense-core amyloid plaques (Gajdusek and Zigas, 1959).
On postmortem, FI is distinguished by thalamic gliosis (astrocyte propagation) but, like GSS, virtually no spongiform damage.
For its part, vCJD exhibits the “florid” plaques depicted in Figure 1: vacuolar lacunae around a core of dense PrP plaques.
Creutzfeldt – Jakob Disease (Varied Phenotypes)
Creutzfeldt-Jakob disease (CJD) is a rare, degenerative, invariably fatal brain disorder. Global prevalence is thought to be just 1 in one million per year worldwide; in the United States, there are about 200 cases per year. CJD usually appears in later life and runs a rapid course. Typically, the onset of symptoms occurs about age 60 and about 90 percent of patients die within 1 year. In the early stages of the disease, patients may have failing memory, behavioral changes, lack of coordination and visual disturbances. As the illness progresses, mental deterioration becomes pronounced and involuntary movements, blindness, weakness of extremities, and coma may occur ( National Institute of Neurological Disorders and Stroke 1).
Sporadic-Type CJD
Sporadic Creutzfeldt – Jakob disease (sCJD) is characterized by the sudden appearance of the infection without any history of gene expression malfunction. Meaning the patient had no known risk factor for the disease when it appears. This type of CJD is the most common and accounts for 85% of all known cases
The first readily identifiable symptoms of sCJD are an abnormal electroencephalogram or EEG, myoclonus, ataxia and rapid degradation of memory, such as confusion and forgetfulness. Symptoms also include blindness, hallucinations, and a speech impediment. People infected by sCJD often experience fatigue, insomnia, vertigo and, logically enough, painful headaches. In the later stages of the disease, ataxia starts to limit the movements of the person to the point of a sudden loss of control over mobility. Falling down and “spaghetti legged-ness” may occur.
As the disease progresses, electroencephalograms show a general pattern of 0.5 to 2.0 Hz against a slow background. A negative EEG can be a “false negative” and does not exclude the disease as not all cases exhibit the typical features. Therefore it is advised that EEGs be done on a weekly basis to capture the abnormality.
By the time gross symptoms present, the disease progresses quite rapidly. During the latter part of the disease, the patient is often bedridden with lingual functions impaired. Often the person afflicted with the disease has a life span of just 5 months to one year. The cause of death is usually sepsis due to incontinence or respiratory complications of the disease.
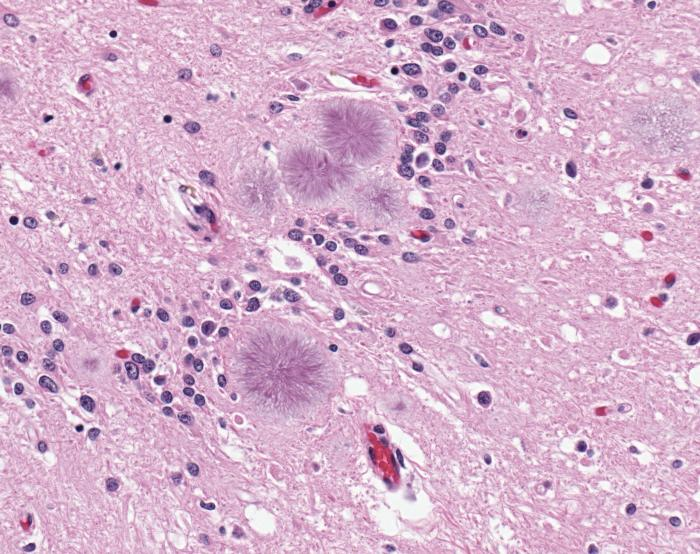
Familial
In familial CJD or “fCJD,” the person affected has a family background of the disease and shows hereditary mutation to genetic makeup. In cases like this, PRNP gene mutation has been inherited by the patient. Spongiform changes occur without GSS-type plaques. Familial CJD accounts for approximately 10% of Creutzfeltd – Jakob Disease cases.
For all their apparent similarity of pathological and clinical phenotypes, sCJD and fCJD have their differences. For instance, persons with fCJD tend to contract the disease 12 years earlier than do patients with sCJD. Another difference is the disease progression lasts 18 months longer than sCJD, lengthening the clinical life of the patient well over a year and up to two.
Iatrogenic CJD
Acquired causes are very rare and account for only 1% of all known CJD cases. Contrary to common belief, as has already been discussed, prion diseases are not airborne nor are persons afflicted with the disease contagious. The only infectious agent of TSEs is the brain and neural tissues. Hence, the only transmission pathways are tissue transplants or medical procedures that transfer vital fluids such as a blood or hGH transfusion.
The first iatrogenic transmission of CJD was reported in the United States after post mortem examination confirmed the diagnosis of the CJD, 18 months after the receipt of a corneal graft. The finding was confirmed when an autopsy revealed that CJD was the cause of death of the cornea donor (Duffy, 1974). Due to the large numbers of corneal grafts and the extended incubation period of CJD, it was reasonable to believe that iatrogenic sporadic CJD will occur in the elderly.
Following observations of the initial iatrogenic transmission of CJD, other reports indicated that CJD transmission in the United States could also occur from exposure to contaminated neurosurgical equipment (Beroulli, 1997), pituitary-growth hormone (hGH) replacement (CDC 1985) and dura mater grafts (CDC, 1987) Elsewhere, there are also reports of similar phenomena from countries like New Zealand, Brazil, UK, Germany and Japan.
The median incubation period for victims with infected hGH exposure was 20.5 years (with a range of 10-30 years) while the median for CJD associated with dura mater grafts was 122 months (range: 14 to 275 months). All the documented cases around the world of hGH exposure were linked to standards for the selection of donors and methods employed in the extraction and purification of hGH (Fradkin, 1991). Likewise, deaths associated with dura mater grafts were linked to minimal oversight and regulation of the procedures for the collection and processing of donor dura mater (Ironsides, 1998).
New Variant CJD
In the late 1980s, a new strain of CJD (vCJD) was reported in younger patients with a median age of 28 years. This was much younger than previously seen with the traditional strain. There was strong evidence to link the apparent variant to BSE and this gained widespread attention as researchers in the UK proved homogeneous patient histories of exposure to BSE tainted beef among a cluster of 20 young patients. Laboratory studies identifying a polymorphic codon 129 of the human prion protein (PrP) gene (PRNP) subsequently strengthened the association between the vCJD and BSE seen in cattle. As of Nov. 2004, 151 cases of vCJD had been reported with an additional 3 cases residing in Canada, Ireland and the USA but likely having been exposed to BSE while in the UK (Belay 2004).
The hypothesized transmission path for BSE lay in the fact that cattle bred for commercial consumption were fed the remains of butchered sheep. Sheep are genetically susceptible to animal-borne prion disease and are the most likely cause for the amplified prion epidemic in Britain. By 1993, more than 1,000 cases of BSE, reported in the popular press as “Mad Cow disease” were being reported on a weekly basis. Before the turn of the century, the morbidity rate had reached 160,000 head of cattle, a prevalence of that amounted to nearly 50% of total herds in the UK. When cases also cropped up on the Continent, several European countries reacted by slaughtering whole herds. However, researchers are still uncertain if the destruction of cattle increased surveillance and discontinued use of offal for cattle feed will prevent a statistically modeled endemic (Ironside 1998).
Sporadic Phenotype: Gerstmann – StrÄUssler – Scheinker Disease
This prion disease was first identified and studied in a German family who suffered from different degrees of pyramidal and extra-pyramidal symptoms, all typical of prion disease. The family also suffered from dementia, dysarthria and ataxia and these became the distinguishing features of GSS disease.
GSS is distinguished from CJD in that EEG discharges are usually absent in the former and on pathological examination, it became known that this phenotype formed plaques around the brain’s cortex that are immunoreactive to PrP antibodies. Patients who contract this disease are usually 50 years and below. The course of this phenotype ranges from 2 to 10 years. Death is not primarily caused by GSS disease but usually comes from a secondary infection, such as pneumonia.
PRNP mutations also typify GSS. A change in the Amino acid coding from proline (P) to leucine (L) at codon 102 was the first mutation of PRNP linked to prion disease. It is the most common GSS-related mutation recognized and has been reported in multiple families from nine different countries, including the original Austrian family described by Gerstmann. This mutation was found to be associated with progressive cerebellar syndrome while another GSS mutation correlates with spinocerebellar degeneration syndrome.
Despite the variation in PRNP mutations, common features among the GSS variants include the absence of periodic discharges on EEG, an early age onset, a moderately prolonged duration averaging 3 years and the presence of GSS plaque pathology,
Familial Phenotype: Fatal Insomnia
Fatal insomnia (FI) is a rare prion disease that causes concern for being autosomal. That is, the causative gene is found in all but the sex chromosomes. Being a dominant trait, it is readily inherited. The dominant gene responsible for its onset has been found in just 50 families worldwide. The probability of offspring inheriting the disease by a newborn child if at least one parent carries the FI gene is at 50%. Like the typical prion disease, once contracted FI progression is irreversible and ultimately leads to death.
The Italian physician Ignazio Roiter stumbled on FI in 1974 because he was mystified by a family where two women had died from insomnia. Upon doing a medical background check on the family Roiter discovered that similar deaths had occurred in the past. The opportunity then presented itself when one of the members of the family contracted the disease. Roiter was able to diagnose and study the disease. Upon the patient’s death, the brain was sent to the United States for further study.
By the late 1990s, researchers had determined that the disease was caused by a dual mutation in a prion protein (the PRNP gene): asparagine-178 replaces aspartic acid while methionine is present at amino acid 129. (Schenkein, 2006). These mutations result in the formation of the insoluble and pathogenic prion protein, PrPSC.
FFI manifests itself between age 30 to 60 and a median age of 50. The early stages of the disease are characterized by increasingly frequent and progressively severe insomnia, panic attacks, and phobia of the paranoid type. As the patient lacks sleep, he begins losing weight rapidly. Four months into frank onset, hallucinations set in. At the terminal stage, dementia also manifests. Little can be done beyond palliative care and addressing the symptoms with psychoactive drugs. Death occurs between 7 and 36 months of onset.
Acquired Phenotype: Kuru
In prior decades, the prion diseases seemed so rare that epidemiologists vaguely thought some “slow virus” was at work. Kuru changed all that in the 1950s when it was recognized as a transmissible pathogen capable of epidemic prevalence. The variant was discovered to be rampant in Papua New Guinea, where health officials documented over 8,000 cases within a single census area of the Okapa subdistrict alone. Field investigation that the main transmission route lay in mortuary cannibalism rituals where mourners literally ate their dead as part of time-honored burial customs. This cannibalism was responsible for the massive spread of the prion protein from person to person. When the practice was outlawed, it seemed that two succeeding generations reached maturity without the prion pathogen (Prusiner 1991). But when it was found in the 1980s and 1990s that kuru had once again reached epidemic proportions and that the median age for onset was 30 years, researchers realized that this acquired phenotype boasted an extended incubation period of 30 to 50 years (Collinge, 2006).
Management of Prion Disease
At the moment, Pharmacology has yet to come up with an answer to battle prion disease, so management of symptoms is presently the sole alternative. For the purpose of delaying PrPsc. production in cell cultures, experimental work has suggested that polysulfated compounds, such as pentosan polysulfate, suramin and heparan sulfate (Diringer, 459; Pocchiari, 560) do work in vitro. Unfortunately, the slowdown effect has been minimal. Another drawback is that the compounds have to be injected before the organism acquires the disease, thus rendering the compounds ineffective in already-infected individuals.
Hence, the most common form of treatment is palliative care. As nothing can be done for the actual progression of the disease, all doctors can do is ease the symptoms of the disease to make the patient more comfortable. Ultimately, chemically induced coma may be applied to reduce the suffering of a terminally-ill patient.
Recent research from the University of Toronto and Caprion Pharmaceuticals has discovered a possible avenue for quicker diagnosis, a vaccine or even a possible cure for the dreaded prion diseases. The abnormal folding proteins which are characteristic of prions and which are behind the onset of the disease have been found to expose a side chain of amino acids which the properly folded protein does not expose. Antibodies that are specifically coded to this side-chain amino acid sequence have been found to stimulate an immune response to the abnormal prions and leave normal proteins intact. (Paramithiotis, 2003)
Other research points to the potential of custom peptide sequences. Apparently, prions congregate by creating beta-barrel structures. In vitro experiments showed the potential of breaking up this clustering by introducing amino acids incompatible with beta barrels.
Further down the road, molecular genetics may provide an answer. Relying on the theory that the gene responsible for protease-resistant protein is an aberrant mutation, researchers will accordingly seek ways to inhibit the gene.
References
Alper, T., Cramp, W. A., Haig, D.A. & Clarke, M. C. (1967). Does the agent of scrapie replicate without nucleic acid? Nature, 214: 764-6.
Alter, H. J. et al. (2007). Emerging infectious diseases that threaten the blood supply. Semin Hematol, 44 (1):32-41.
Aquzzi, A. & Glatzel, M. (2006). Prion infections, blood and transfusions. Nat Clin Pract Neurol., 2 (6): 321-329.
Barletta, J. (2006). Applications of real-time immuno-polymerase chain reaction (rt-IPCR) for the rapid diagnoses of viral antigens and pathologic proteins. Mol Aspects Med, 27 (2-3): 224-253.
Belay, E. D. et al. (2004a) Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease. Neurology, 60: 176-181.
Belay, E. D. et al. (2004b) Chronic Wasting Disease and potential transmission to humans. CDC, Emerging Infect Dis. Web.
Bernolli, C. et al. (1977). Dangers of accidental person-to-person transmission of Creutzfeldt-Jacob disease by surgery. Lancet, (1):478-479.
Bren, L. (2001). Trying to keep “mad cow disease” out of U.S. herds. FDA Consum, 35 (2):12-14.
Brown, P., Cathala, F., Raubertas, R. F., Gajdusek, D. C. & Castaigne, P. (1987). The epidemiology of Creutzfeldt-Jakob Disease: Conclusion of a 15-year investigation in France and review of the world literature. Neurology, 27: 895-904.
Brown, P. et al. (2003) WHO manual for surveillance of human transmissible spongiform encephalopathies, including variant Creutzfeldt – Jakob disease. Geneva: World Health Organization.
Cai, L. et al. (2005). Ensuring the biologic safety of plasma-derived therapeutic proteins: Detection, inactivation and removal of pathogens. BioDrugs, 19 (2):79-96.
Center for Disease Control and Prevention (1985). Fatal degenerative neuralgic disease in patients who received pituitary-derived human growth hormone. MMWR 34: 359-360, 365-366.
Center for Disease Control and Prevention (1987). Rapid progressive dementia in a patient who received a cadaveric dura matter graft. MMWP 36: 49-50 and 55.
Center for Disease Control and Prevention (2008). File: Variant Creutzfeldt-Jakob disease (vCJD), typical amyloid plaques, H&E.jpg
Colee, J. G. et al, (1997). BES: A decade on- Part I. Lancet, 349: 715-721.
Collinge, J. et al. (2006). Kuru in the 21st century – An acquired human prion disease with very long incubation periods. Lancet, 367 (9528):2068-2074.
Collinge, J. (2001). Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci, 24: 519–50.
Concepcion, G.P. & Padlan, E. A. (2005). The codon for methionine at position 129 (M129) in the human prion protein provides an alternative initiation site for translation and renders individuals homozygous for M129 more susceptible to prion disease. Med Hypotheses, 65 (5): 869-867.
Cuillé, J. & Chelle, P. L. (1939). Experimental transmission of trembling to the goat. C R Seances Acad Sci, 208: 10-58-60.
De Hayen, W. R., et al. (2006). Animal health: Foundation of a safe, secure and abundant food supply. J Vet Med Educ, 33 (4): 496-501.
Dringer, H. & Elers, B. (1991). Chemophrophylaxis of scrapie in mice. J Gen Virol, 72: 457-460
Duffy P. et al. (1974). Possible person-to-person transmission of Creutzfeldt-Jakob disease. N. Engl. J. Med., 290:692-693.
Elbourn, R. E. (1999) The prion diseases: A molecular and genetic perspective. Unpublished Diss. Web.
Gajdusek, D. C., Gibbs, C. J., Jr. & Alpers, M. (1966). Experimental transmission of a kuru-like syndrome to chimpanzees. Nature, 209: 294-6.
Gajdusek, D. C. & Zigas, V. (1959). Clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the eastern highlands of New Guinea. Am J Med, 26: 442-469.
Giaccone, G., Tagliavini, F., Verga, L. et al. (1990). Neurofibrillary tangles of the Indiana kindred of the Gerstmann-Sträussler-Scheinker disease share antigenic determinants with those of Alzheimer disease. Brain Res, 530: 325-9.
Goodnough, L. T. et al. (2004) Transfusion medicine review – Joint ASH and AABB educational session. Hematology. The American Society of Hematology.
Gregori, L. et al. (2004). Effectiveness of leucoreduction for the removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet; 364: 529-531.
Georgsson, G. et al. (2006). Polymorphism of PRNP codons in the normal Icelandic population. Acta Neurol Scan, 113 (6):419-425.
Hewitt, P. E. et al. (2006). Creutzfeldt-Jakob disease and blood transfusion: Results of the UK Transfusion Medicine Epidemiological Review Study. Vox Sang, 91 (3):221-230.
Hsiao, K., Cass, C. Conneally, P. M. et al. (1990). Atypical Gerstmann-Sträussler-Scheinker syndrome with neurofibrillary tangles: No mutation in the prion protein open-reading frame in a patient of the Indiana kindred. Neurobiol Aging, 11: 302.
Ironside, J. W. (1998). Neuropathological findings in new variant CJD and experimental transmission of BSE. FEMS Immunol. Med. Microbiol., 21:91-95.
Konold, T. et al. (2006). Different prion disease phenotypes result from inoculation of cattle with two temporally separated sources of sheep Scrapie from Great Britain. BMC Vet Res, 17:2-31.
Llewelyn, C. A., et al. (2004). Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet, 363:411-412.
MacGregor, I. R. & Prowse, C.V. (2004). Impacts and concerns for vCJD in blood transfusion: Current status. Curr Mol Med, 4:361-373.
Masters, C. L., et al. (1979). Creutzfeldt-Jacob disease: Patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol, 5:177-188.
Mastrianni, J. A., M.D., Ph.D. & Roos, R. P., M.D., (2000). The Prion diseases. Seminars in Neurology. 20 (3):337-52.
McKinley, M. P., Bolton, D. C. & Prusiner, S. B. (1983). A protease-resistant protein is a structural component of the scrapie prion. Cell, 35: 57-62.
National Institute of Neurological Disorders and Stroke (2009) Creutzfeldt-Jakob disease fact sheet. National Institutes of Health.
Paramithiotis, E., Pinard, M., Lawton, T., et al. (2003). A prion protein epitope selective for the pathologically misfolded conformation. Nature Medicine, 9 (7): 893–9.
Peden, A. H., et al. (2004) Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet, 364: 527-529.
Pocchiari, M., Salvatore, M. & Ladogana, A., et al. (1991). Experimental drug treatment of scrapie: A pathogenetic basis for rationale therapeutics. Eur J Epidemiol, 7: 556-561.
Prowse, C. V., MacGregor, I. R. (2002). Mad cows and Englishmen: An update on blood and vCJD. Var Sang, 83:341-349.
Prusiner, S.B., McKinley, M. P., Groth, D.F. et al. (1981). Scrapie agent contains hydrophobic protein. Proc Natl Acad Sci U.S.A., 78: 6675-6679.
Prusiner, S.B. (1982). Novel proteinaceous infectious particles cause scrapie. Science, 216 (4542): 136–44.
Prusiner, S.B. (1991) Molecular biology of prion diseases. Science, 252: 1511-1521.
Public Health Agency of Canada (2006). Creutzfeldt-Jakob Disease. Web.
Qin, K.; et al. (2006). Doppel: More than double to prion. Neurosciences, 141 (1): 1-8.
Safar, J., Roller, P. P., Gajdusek, D. C. & Gibbs, C. J. Jr. (1993). Conformational transitions, dissociation and unfolding of scrapie amyloid (prion) protein. J Biol Chem, 268: 20276-20284.
Schenkein, J. & Montagna, P. (2006). Self management of fatal familial insomnia. Part 1: What is FFI? Medscape General Medicine, 8 (3).
Sigurdsson, B. (1954). Rida, a chronic encephalitis of sheep with general remarks on infections which develop slowly and some of their special characteristics. Br. Vet. J, 110: 341-354.
Vollmert C., et al. (2006). Significant association of a M129 independent polymorphism in the 5’ UTR of the PRNP gene with sporadic Creutzfeldt-Jakob Disease in a large German case-control study. J Med Genet, 43 (10): e53.
Warwick, R. M. et al. (2005). Should decreased donors be tested for vCJD? Cell Tissue Bank, 6 (4):263-270.
Wickner, R.B., et al. (2004). Prion genetics: New rules for a new kind of gene. Annu Rev Genet, 38:681-707.
Will, R. G. (1993). Epidemiology of Creutzfeldt-Jakob disease. British Med Bulletin, 49 (4): 960-970.