Introduction
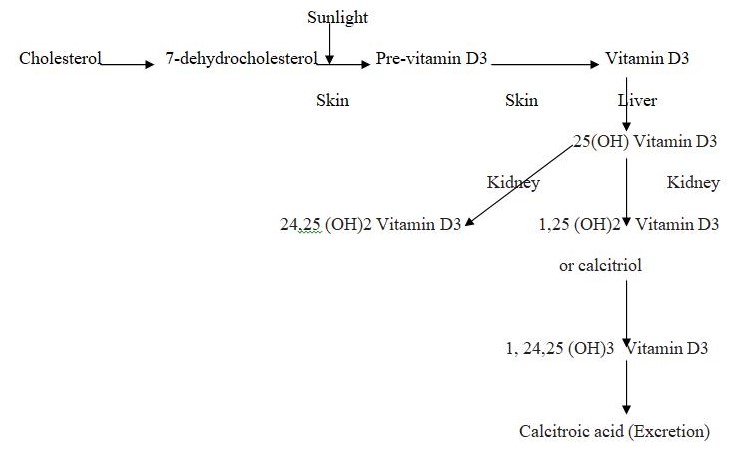
The skin is the barrier between the human body and the environment. Its functions are to prevent microorganisms from entering the body, ultra-violet radiation from affecting the body and water loss from the body (Ravid, 2002, p 525). The continuous regeneration of the terminally differentiating keratinocytes constitutes the barrier. The complete ultraviolet-B (UV-B)-induced pathway from 7-dehydrocholesterol to hormonally active form of Vitamin D called 1, 25-dihydroxyvitaminD3 or calcitriol under physiological conditions occurs only in the skin in the epidermal cells or keratinocytes (Lehmann, 2005, p. 1246). Calcitriol synthesis is of significance as it regulates important cell functions in the keratinocytes and immunocompetent cells. The keratinocytes also have the nuclear Vitamin D receptor which mediates the hormonal effects. The skin exposure to uv radiation through excess sunlight is harmful in that skin cancers can result but proves beneficial in that the energy obtained as radiation may be used to convert 7-dehydrocholesterol to pre vitamin D3 which is then thermally isomerised to Vitamin D3 or calcitriol (Bikle, 2004, p 3472S)
Background
Significance of Vitamin D to human cancers
Skin cancer is the most common cancer affecting humans especially the fair-skinned.
The skin consists of the epidermis made up of squamous epithelial cells and keratinocytes while the dermis consists mainly of fibroblasts (D’Errico, 2006, p. 37). The epidermis is four-layered. Melanomas and carcinomas affecting the keratinocytes are the cancers. Squamous epithelial carcinomas arise from the epithelial cells and Basal cell carcinomas from the keratinocytes at the basal layer of cells of the epidermis.
The biologically active calcitriol prevents the development of malignancies by stimulating the differentiation of the cells in a number of mechanisms which have been proved mostly in vitro on rodents (Bikle, 2004, p. 3472S). Any disturbance that occurs in the differentiation or in the prevention of proliferation leads to cancers. Calcium and Vitamin D3 together regulate the differentiation process of the cell in vivo (Bikel, 2004, p. 3476S). A persistent rise in calcium ions is the first requirement. Vitamin D3 activates and induces the phopholipase C and the opening of calcium channels. It also acts on the genes which produce the proteins for differentiation. Further it regulates the Vitamin D receptor which binds to one of two coactivator complexes: DRIP or p160/SRC. The DRIP is activated when the keratinocyte is undifferentiated. As differentiation occurs through maturity, the other complex is activated and the differentiation is completed. Many agents have been identified for the differentiation and inhibition of proliferation processes. The gradient of Calcium, highest in the outer cells of the epidermis and lowest in the innermost basal layer achieves the two processes. The calcitriol is a calcium-regulating hormone which therefore protects the keratinocytes from damage (Mason and Holliday, 2000).
Human beings are exposed to harmful or genotoxic and beneficial or antioxidant chemicals. Hazardous exposures include smoking and sun tanning or inadequate intake of vegetables and fruits which are cancer-protective (Moller, 2006, p. 336). When the exposed individuals become more in number, risk assessment becomes important.
Treatment with Vitamin D causes envelope formation in keratinocytes (Holliday et al, 1997) and increased melanogenesis in melanocytes (Ranson et al, 1988). Dissanayake et al confirmed this (1993). The active Vitamin D hormone that is synthesized in the skin following uvr increases the survival of skin cells and the DNA damage too would be minimal (Gupta, 2007, p. 207). Survival of keratinocytes was therefore better. The thymine dimmers were also reduced in the surviving cells. Nuclear p53 expression increased especially in the cells with active Vitamin D (Gupta, 2007, p. 207). Number of sunburn cells were seen reduced in the skin of Skh:hr1 mice 24 hours after being treated with active Vitamin D. Gupta’s findings are consistent with the proposal that the Vitamin D system forms a part of the protective mechanism against the damage caused by uvr.
The metabolic changes of the vitamin D3 is shown in Figure 1. A photochemical reaction converts the 7-dehyrocholesterol to Vitamin D3 which is converted to 1,25-dihydroxycholecalciferol either in the liver or the keratinocyte itself. The parathyroid hormone stimulates the production (Bikle, 2004, p. 3473S). An investigation in the HaCaT keratinocytes confirmed that Previtamin D3, time-dependent thermal isomerisation to Vitamin D3 and simultaneous formation of calcitriol or 1, 25-dihydroxycholecalciferol all occur with the UVB rays (Lehmann, 2000, p. 803). It can be understood that keratinocytes could be a potential source of calcitriol when irradiated with therapeutic doses of UVB. The chain of reactions include photochemical VD3 synthesis in the skin and subsequent hydroxylation at the C-25 atom in the liver and at C-1a position in the kidney. Keratinocytes of the HaCaT cell line convert exogenous VD3 to calcitriol (Boukamp, 1988).
DNA damage and repair mechanisms
Exposure to sunlight allows ultra-violet radiation (UVB) of wavelength 290-320 nm to penetrate the epidermis and cause alterations in the structure of DNA which could produce mutations or carcinogenesis, both harmful effects (Chevalier, 1997, p. 325). The damage has been attributed to thymine dimer formation (Setlow, 1966 as cited in Gupta, 2007, p. 207). The p53 tumour suppression protein is increased through post translational stabilisation (Hall et al, 1993). This activates the process by which cell cycle is arrested. The increased p53 causes the
DNA repair systems to become mobilized automatically (Smith et al, 1995). p53 can also cause apoptosis when there is excessive unrepaired DNA damage (Decraene et al, 2001). A finding has been reported where mice treated with Vitamin D reduced nitric oxide and apoptosis (Riachy et al, 2002). DNA repair prevents the mutations and carcinogenesis. The repair could occur as a gene expression which controls the nucleotide repair mechanism. Proteins like p53 may come into action in the role of a transcription factor to control the cell cycle progression or initiation of the apoptosis mechanism (Yarosh, 2002, p. 58).
Irradiation by sunlight exposure could lead to alterations in the process which could form cytotoxic photoproducts (Chevalier, 1997, p.326). Greater exposure would produce excessive concentration of Vitamin D3. The DNA repair system would operate to eliminate the cells which are most potent to produce mutations and carcinogenesis. Vitamin D3 concentration in the cell is reduced by the passing into the circulation and could be understood as part of the inactivation and excretion. This indicates that 7-DHC could be a potential means of treatment of proliferative disease. Also the use of sunscreens with a UVB filter could prove useful in limiting the formation of mutations (Chevalier, 1997, p. 326). The base excision repair is involved in most repair mechanisms in tissues (Hegde, 2008, p. 28). Genomic integrity is maintained by this evolutionary derived process.
UV Radiation
UVB UV radiation is the most significant environmental factor in the production of skin cancers (Assefa, 2000, p. 21416). Of the fractions, only UVB irradiations have contributed to carcinogenesis. The potentially dangerous UVC is absorbed by the ozone layer and never reaches the earth. “UVB-induced apoptosis in keratinocytes has been shown to be significantly counteracted by inhibitors of the initiator caspase-8 such as cytokine response modifier A and zVAD-fmk, or by the expression of dominant negative FADD” (Argane, 1998). The exposure of keratinocytes to UVB triggers the expression of many genes which are involved in various processes like cell cycle arrest, DNA repair and /or apoptosis. Apoptosis is believed to be a protective mechanism for removing irreversibly damaged cells due to cancer. Cytosolic and nuclear events are related to apoptotic response. Whether cells undergo apoptosis upon UVB irradiation is the deciding factor for photocarcinogenesis (Assefa, 2000, p. 21416). Some studies have shown that exposure to UVB irradiation induces the activation of “p38 MAPK, cytochrome c release, pro-caspase-8 and -3 activation, Bid cleavage and apoptosis in HaCaT cells” (Assefa, 2000, p.21419). Skin cancers are usually developed due to the induction of DNA damage by excessive solar UVB radiation (Courdavault, 2005, p. 836). It is known that UVB only maintains two types of DNA damage: cyclobutane pyrimidine dimmers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (64 PPs) (Courdavault, 2005, p.210). Evidence for this is supported by the large number of mutations occurring at the bipyrimidine sites at tumors (Courdavault, 2005, p. 836).
UVA UVA radiation (320-400nm) which is less energetic but occurs in abundance also shows mutagenic properties even though poorly absorbed. The action of UVA is believed to be indirect through the cellular chromophores by inducing photo-oxidative cellular stress (Wondrak et al, 2006). When the endogenous chromophores are excited, energy is transferred to oxygen to yield reactive oxygen that reacts with the guanine bases of DNA. Direct oxidation also could occur. The reactive oxygen species generate UVA signature mutations by which transverse mutations occur converting guanine to thymine (Cadet et al, 2005). Studies have shown that UVA causes the formation of CPDs, cyclobutane pyrimidine dimers. Skin cells and rodent cells have shown that CPDs were found more than the guanine product (Courdavault, 2004). Long lasting DNA strand breaks are caused by physiologically relevant doses of UVA radiation which forms the major portion of UV radiation. This leads to chromosomal aberrations and cause the HaCaT cells to become tumours (Wischerman, 2008, p. 4269). UVA is therefore also gaining significance in the contribution to skin cancers. In vitro studies in albino hairless mice showed skin tumours about 400 days after UVA radiation. Human HaCaT keratinocytes were found to show tumours when irradiated with UVA for 18weeks. (He et al, 2006).
p53
p53 when elevated induces the cell cycle arrest, causes the DNA repair and directly leads to the increased nucleotide excision repair following uvr (Smith et al, 1995). In the presence of excessive DNA damage, the increased p53 can cause apoptosis by upregulation of Bax or downregulation of Bcl-2 (Ouhtit et al, 2000). Treating with vitamin D did not produce the expression of p53 in nonirradiated cells (Sebag et al, 1994). Studies have shown that UVB induces mutations which are unique in the p53 tumour suppressor gene. Other carcinogens do not show this (Benjamin, 2007, p. 55).
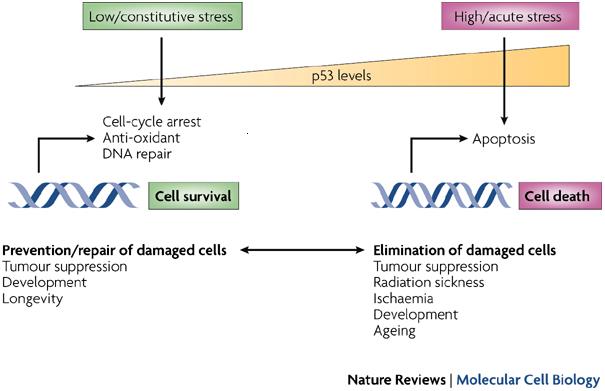
Studies on mice have shown that p53 mutations were very high. These mutations were seen before the appearance of tumours. It has been suggested that the efficiency of suncreens could be tested using the appearance of p53 mutations as biological endpoints. As p53 mutations are early incidents in photocarcinogenesis, their detection and inhibition of the incident would protect against the development of skin cancer (Benjamin, 2007, p.55). p53 is a cellular protein. As the wild type, it is a tumour suppressor or anti-oncogene. Normal growth is maintained by it. It also produces genomic stability by causing a cell cycle arrest or inducing apoptosis in response to stressors (Vogelstein, 1992). The cell cycle arrest allows much of the cell repair pathways to remove most of the DNA lesions before DNA synthesis and mitosis starts.
Apoptosis
Vitamin D is believed to act on tumours by enhancing the susceptibility to apoptosis-inducing agents (Koren et al, 2000). Apoptosis is a program for cell death caused by the drugs used in the treatment of malignant diseases (Darzynkiewicz, 1995). It can occur through the uvr induction directly and due to the expression of p53 (Gupta, 2007, p. 711). When solar uv irradiation damages the skin cells, the cells are initiated and grow into tumours. Hyperproliferation occurs and cancerous cells are formed through clonal expansion and apoptosis evasion (Reagan-Shaw, 2006, p.418). Cancer development can be prevented by chemopreventive agents which prevent the formation of the initiated cells or eliminate the potentially dangerous initiated cells. Sanguinarine has been suggested as one such chemopreventive agent. It is believed to protect skin cells from the apoptotic elimination of damaged cells in DNA damage which do not undergo the programmed cell death and the process may be through clonal expansion (Reagan-Shaw, 2006, p. 418).
The cellular pathways that lead to apoptosis are yet to be understood better so that chemotherapy is improved. Studies are being done extensively to reach the answers early. The activation of the executioner process which causes apoptosis is linked to the release of mitochondrial cytochrome C (Wang, 1997, p.210). The release is inversely related to Bcl2 and Bcl-XL proteins (Wang, 1997, p. 210). Another cellular anti-apoptotic protein Mcl-l showed increases in another study in which human leukemia cells were treated with calcitriol. There was also an increase in A1 protein level but no increase in Bcl-2 and Bcl-XL proteins. The Bcl-2 family members have survival enhancing and pro-apoptotic properties (Wang, 1997, p.210).
A study showed that the activated vitamin D3, calcitriol, did not induce apoptosis through the formation of ceramides as was believed by some researchers. In fact it was found to protect the keratinocyte by resisting the apoptosis facilitated by the ceramides or uv irradiation or the tumor necrosis factor alpha (Manggau, 2001, p. 1241). The vitamin D3 was found to protect the cells from apoptosis by mediating through sphingosine1 phsophate (Manggau, 2001. p.1241)
The comet assay “The comet assay is a rapid method used in genotoxicity testing in vitro and in vivo and in biomonitoring studies for detecting low levels of DNA damage”(Tice, 2000). More information on the type of DNA damage may be obtained. The significant event in carcinogenesis is the generation of DNA damage (Moller, 2006, p. 336). The comet assay is useful in biomonitoring. DNA migration characterized by “strand breaks, alkaline labile sites and transient repair sites may be detected by a simple alkaline comet assay” (Moller, 2006, p. 336).
Advanced DNA migration including oxidative damage, alkylations and UV light-induced photoprotective products are detected after incubation with endonucleses. The lysed cells are incubated with lesion-specific endonucleases to improve sensitivity and specificity. The endonucleases are many in number and produce different manners of DNA repair. Endonuclease III can recognise different types of oxidative damage (Smith, 2006, p. 185). The alkaline comet assay is done following radiation and recovery of the cells in a complete culture medium (Wischermann, 2007). All steps are executed on ice to reduce the repair processes.
The alkaline comet assay is used to detect genotopic exposure in humans. Risk assessment may require the use of the comet assay for defining hazards. Sufficient validation would allow its being used for risk assessment. Early biological defects are biomarked by chromosomal aberration and micronucleus frequency. The comet assay is an exposure assay which imparts information on the biologically effective dose. Biomarkers must be thoroughly validated for application as tools for risk assessment (Moller, 2006, p. 336). The validation of biomarkers involves four phases of laboratory studies, transitional studies, aetiologic studies and public health applications.
The comet assay was developed as a method to detect DNA damage in the 1980s. The main uses of the comet assay have been in the detection of the “DNA-DNA crosslinks and gene specific DNA damage” (Moller, 2006, p. 339). Repair activity in cellular extracts has been detected by a modified procedure. Enzyme digestion with DNA glycosylase or endonuclease enzymes helps detect the oxidized pyrimidines and purines of further DNA damage. One of the most important lesions detected in this manner is the “premutagenic 7-hydro-8-oxo-2o-deoxyguanosine lesion” (Moller, 2006, p. 339).
Study purpose
This study aims to investigate whether 1, 25(OH)2D3 has a protective effect on total DNA damage, T<>T and oxidative damage, using the alkaline comet assay. This will be done in cell culture using human keratinocytes and a solar simulator. Investigation of post-irradiation apoptosis and cell survival are also being done.
Significance of study
The commonest cancers occurring in human beings are the skin cancers. Exposure to sunlight or uv irradiation makes skin prone to cancers arising from the squamous cells, basal cells or melanin cells of the epidermis. Previous studies have indicated that the exposure to uv irradiation converts the 7 dehydrocholesterol in the skin to 1, 25 dihroxyvitamin D3 or calcitriol which is the biologically active form. Normally this form facilitates the anti-proliferative and prodifferentiative functions which prevent the arising of malignancies. The normal repair processes in the keratinocytes of the skin occur in the presence of the calcitriol whereby the excess and damaged cells are removed. Any disturbance of the cell functions and repair processes by way of excessive exposure to sunlight could produce the tumours of the skin.
Studies have found that UVB and UVA are harmful to the skin cells. The photoprotective effects of 1.25 (OH) 2D3 have been mentioned in previous studies. This study intends to explore the effects in detail and post-irradiation apoptosis and cell survival in order to secure more information as to how the 1.25 (OH) 2D3 can protect the skin keratinocytes from total damage, mutation and carcinogensis
Research Hypotheses
- The 1.25 (OH) 2D3 or calcitriol has a protective action on the keratinocytes by preveting mutations , carcinogenesis and total damage.
- The post- irradiation apoptosis is sufficient as repair for the keratoncytes.
- Cell survival is determined by the 1.25 (OH) 2D3.
Limitations
This study is being conducted in animals so the conditions for testing may not suit human cells. Conclusions can be made only tentatively and cannot be immediately applied to humans.
Assumptions
This study is being done as one more among the various researches being done on the topic. Some more information will be obtained and this maybe compared to the findings of recent studies.
Summary
The photoprotective effects of the skin which is exposed to ultraviolet irradiations has been discussed in this chapter. The uvb portion of the uvr produces maximum effects on skin. However now even the uva portion appears to produce changes in the skin. The uvr converts 7 dehyrdo cholesterol to the biologically active 1,25 dihydroxycholecalciferol or calcitriol. This hormone has anti-proliferative and prodifferentiative functions in the keratinocytes. Normally this hormone functions so that the DNA damage that has occurred is repaired via various mechanisms which would ensure a normal cell cycle and prevention of tumour formation. However changes like excessive exposure to uvr leads to excessive active Vitamin D3 formation and exaggeration of all functions. Even in the presence of excessive exposure the cells respond well so that no lesions occur. However when the system becomes faulty, damage is more than repair and proliferation of cells occur leading to skin cancers. Studies have shown various manners in which the skin protects itself from dysfunction and tumour formation. How we can diagnose early skin cancer even before the tumour presents itself has been suggested by some studies. The effects of sunscreen lotions and how they can best act has been suggested by other studies using the p53 protein. Various methods are available but the alkaline comet assay has been selected for the study.
References
- Aragane, Y., Kulms, D., Metze, D., Wilkes, G., Poppelmann, B., Luger, T. A., and Schwarz, T. (1998) J. Cell Biol. 140, 171–182
- Assefa, (2000) “p38 Mitogen-activated Protein Kinase Regulates a Novel, Caspase-independent Pathway for the Mitochondrial Cytochrome c Release in Ultraviolet B Radiation-induced Apoptosis THE JOURNAL OF BIOLOGICAL CHEMISTRY. Vol. 275, No. 28, pp. 21416–21421 The American Society for Biochemistry and Molecular Biology, Inc.
- Benjamin, C.L. (2008). p53 Tumor Suppressor Gene: A Critical Molecular Target for UV Induction and Prevention of Skin Cancer. Photochemistry and Photobiology, 2008, 84: 55–62
- Bikle, D. (2004). Vitamin D and Skin Cancer Journal of Nutrition. 134: 3472S–3478S, 2004. American Society for Nutritional Sciences.
- Boukamp, P., R. T. Petrussevska, D. Breitcreutz, J. Hornung, A. Marham and N. E. Fusenig (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell. Biol. 106, 761–771.
- Cadet J, Sage E, Douki T. (2005). Ultraviolet radiation-mediated damage to cellular DNA. Mutat Res 571: 3–17.
- Chevalier, G. et al. (1997). Was the formation of 1,25 dihydroxyvitamin D3 initially a catabolic pathway. Medical hypotheses, Vol. 48, p 325-329. Pearson professional Limited
- Courdavault, S., C. Baudouin, M. Charveron, A. Favier, J. Cadet, T. Douki, Larger yield of cyclobutane dimers than 8-oxo-7,8-hydroguanine in the DNA of UVA-irradiated human skin cells, Mutation Res. 556 (2004) 135–142.
- Darzynkiewicz, Z. J. (1995) Cell. Biochem. 58, 151–159.
- Decraene D, Agostinis P, Pupe A, de Haes P, Garmyn M (2001) Acute response of human skin to solar radiation: regulation and function of the p53 protein. J Photochem Photobiol B 63:78–83
- Derrico, M et al. (2006). Cell type and DNA damage specific response of human skin cells to environmental agents. Mutation Research 614 (2007) 37–47, ScienceDirect
- Dissanayake NS, Greenoak GE, Mason RS (1993) Effects of ultraviolet irradiation on human skin-derived epidermal cells in vitro. J Cell Physiol 157:119–27
- Gupta, R. et al (2007). Photoprotection by 1,25 Dihydroxyvitamin D3 Is Associated with an Increase in p53 and a Decrease in Nitric Oxide Products Journal of Investigative Dermatology (2007) 127, 707–715. doi:10.1038/sj.jid.5700597;
- Hall PA, McKee PH, Menage HD, Dover R, Lane DP (1993) High levels of p53 protein in UV-irradiated normal human skin. Oncogene 8:203–7
- Hegde, M.L. et al. (2008). Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells, Cell Research (2008) 18:27-47. IBCB, SIBS, CAS
- He YY, Pi J, Huang JL, Diwan BA, Waalkes MP, Chignell CF. (2006). Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance. Oncogene 25: 3680–3688.
- Holliday C, Lee JK, Posner GH, Mason RS (1997) Bioactivity of hybrid analogs of 1,25 dihydroxyvitamin D3 in skin cells. In: Vitamin D, chemistry, biology and clinical applications of the steroid hormone. (Norman A, Bouillon R, and Thomasset M. eds), Riverside: University of California, Printing and Reprographics, 95–6
- Hosomi, J., J. Hosoi, E. Abe, T. Suda and T. Kuroki (1983) Regulation of terminal differentiation of cultured mouse epidermal cells by 1alpha ,25-dihydroxyvitamin D3. Endocrinology 113, 1950–1957.
- Koren R, Rocker D, Kotestiano O, Liberman UA, Ravid A (2000) Synergistic anticancer activity of 1,25-dihydroxyvitamin D(3) and immune cytokines: the involvement of reactive oxygen species. J Steroid Biochem Mol Biol 73:105–12
- Lehmann B, Rudolph T, Pietzsch J & Meurer M (2000). Conversion ofvitamin D3 to 1 alpha,25-dihydroxyvitamin D3 in human skin equivalents. Experimental Dermatology 9 97–103.
- Lehmann, B. (2005). UV Radiation, Vitamin D and Human Health: An Unfolding Controversy The Vitamin D3 Pathway in Human Skin and its Role for Regulation of Biological Processes. Photochemistry and Photobiology, Vol. 81. American Society for Photobiology
- Manggau, M. et al (2001). 1a,25-Dihydroxyvitamin D3 Protects Human Keratinocytes from Apoptosis by the Formation of Sphingosine-1-Phosphate. J Invest Dermatol 117:1241-1249, 2001
- Mason RS & Holliday CJ 2000 1,25-Dihydroxyvitamin D contributes to photoprotection in skin cells. In Vitamin D Endocrine System. Structural, Biological, Genetic and Clinical Aspects, pp 605–608. Eds AW Norman, R Bouillon & M Thomasset. Riverside: University of California.
- Moller, P. (2006). The Alkaline Comet Assay: Towards Validation in Biomonitoring of DNA Damaging Exposures, Basic and Clinical Pharmacology and Toxicology, Vol. 98, P.336-345
- Ouhtit A, Muller HK, Davis DW, Ullrich SE, McConkey D, Ananthaswamy HN (2000) Temporal events in skin injury and the early adaptive responses in ultraviolet-irradiated mouse skin. Am J Pathol 6:201–7
- Ranson M, Posen S, Mason RS (1988) Human melanocytes as a target tissue for hormones: in vitro studies with 1 alpha-25, dihydroxyvitamin D3, alpha-melanocyte stimulating hormone, and beta-estradiol. J Invest Dermatol 91:593–8
- Ravid, A. et al. (2002). Vitamin D inhibits the activation of stress-activated protein kinases
- by physiological and environmental stresses in keratinocytesJournal of Endocrinology (2002) 173, 525–532 0022–0795/02/0173–525 Society for Endocrinology Printed in Great Britain
- Reagan-Shaw, S. et al, (2006). Enhancement of UVB radiation–mediated apoptosis by sanguinarine in HaCaT human immortalized keratinocytes. Mol Cancer Ther 2006;5(2):418–29]
- Riachy R, Vandewalle B, Kerr Conte J, Moerman E, Sacchetti P, Lukowiak B et al. (2002) 1,25-Dihydroxyvitamin D3 protects RINm5F and human islet cells against cytokine-induced apoptosis: implication of the antiapoptotic protein A20. Endocrinology 143:4809–19
- Sebag M, Gulliver W, Kremer R (1994) Effect of 1,25 dihydroxyvitamin D3 and calcium on growth and differentiation and on c-fos and p53 gene expression in normal human keratinocytes. J Invest Dermatol 103:323–9
- Smith ML, Chen IT, Zhan Q, O’Connor PM, Fornace AJ Jr (1995) Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene 10:1053–9
- Smith, C.C. (2006). hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis vol. 21 no. 3 pp. 185–190, 2006, The Author , 2006
- Tice,R.R., Agurell,E., Anderson,D., Burlinson,B., Hartmann,A.,Kobayashi,H., Miyamae,Y., Rojas,A., Rye,J.-C. and Sasaki,Y.F. (2000) Single cell Gel/Comet assay: guidelines for in vivo and in vitro genetic toxicology testing. Environ. Mol. Mutagen., 35, 206–221.
- Vogelstein, B. and K. W. Kinzler (1992) p53 function and dysfunction. Cell 70, 523–526.
- Vousden, K. H. and Lane, D. P. (2007) Nature Reviews Molecular Cell Biology 8, 275-283 (2007) doi:10.1038/nrm2147
- Wang, X. et al. (1997). Antiapoptotic Action of 1,25-Dihydroxyvitamin D3 Is Associated with Increased Mitochondrial MCL-1 and RAF-1 Proteins and Reduced Release of Cytochrome c. EXPERIMENTAL CELL RESEARCH 235, 210–217
- Wischermann, K. et al (2008). UVA radiation causes DNA strand breaks, chromosomal Aberrations and tumorigenic transformation in HaCaT skin keratinocytes Oncogene (2008) 27, 4269–4280 Macmillan Publishers Limited
- Wischermann K, Boukamp P, Schmezer P. (2007). Improved alkaline comet assay protocol for adherent HaCaT keratinocytes to study UVA-induced DNA Damage. Mutat Res 630: 122–128.
- Wondrak GT, Jacobson MK, Jacobson EL. (2006). Endogenous UVAphotosensitizers: mediators of skin photodamage and novel targets for skin photoprotection. Photochem Photobiol Sci 5: 215–237.
- Yarosh, D.B. et al, (2002). Measurement of UVB-Induced DNA damage and its consequences in models of immunosuppression Methods 28 (2002) 55–62 Academic Press.