Introduction
Glucose 6 phosphate dehydrogenase (G6PD) is a protein enzyme found in the cytosol of human and bacterial cells. It exists as a monomer made up of 515 amino acids and a molecular weight of 59 kDa. (Glew & Rosenthall, 2009) It is found in all cells but its concentration in different tissues varies. In humans specifically, it is the first enzyme of the alternative pathway in glucose metabolism i.e. the pentose phosphate pathway. (Vanella et al) The chief function of this pathway is the production of nicotinamide adenine dinucleotide phosphate (NADPH) for steroid and fatty acid synthesis and also produce ribose required for nucleic acid biosynthesis. (Smith, 1979) It is activated by either its substrate glucose 6 phosphate or by insulin as it acts to reduce the blood sugar levels.
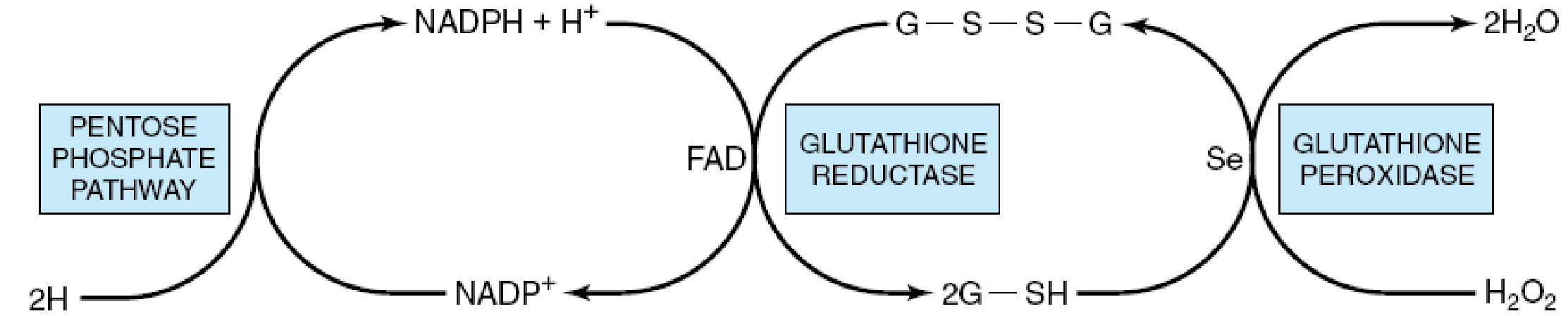
Role of the pentose phosphate pathway in the glutathione peroxidase reaction of erythrocytes. (G-S-S-G, oxidized glutathione; G-SH, reduced glutathione; Se, selenium cofactor.)
From the foregoing, a conclusion was arrived at that a deficiency in the G6PD enzyme will lead to an increase in cellular susceptibility to oxidative stress, especially erythrocytes, that are utterly dependent on the pentose pathway as they do not have mitochondria for NADPH production to survive the stress. (Mengel, Frei & Nachman, 1972)
This paper is intended to highlight what is known about glucose 6 phosphate dehydrogenase deficiency and its relation to haemolytic anaemia and its relation to haemolytic anaemia. To prove experimentally that the deficiency is a cause of the haemolysis by simulating an actual variant of G6PD deficiency and subject it to the ideal oxidative stress that ordinary red blood cells undergo. Thereafter, a discussion of the possible mechanism behind the haemolysis and conclusion.
Glucose 6 Phosphate Dehydrogenase Deficiency
Glucose 6 phosphate dehydrogenase deficiency has been established as a clinical disorder and defined as a hereditary defect in the gene coding for it i.e. the G6PD gene. According to (Motulsky & Yoshida, 1969) it is one of the most commonly known disorders that are inheritable in man. It is X- linked and the mutation results in different proteins with altered enzymatic activity and thus associated with clinically and biochemically different phenotypes. Its advent dates back to the 20th century when eating fava beans and taking of the antimalarial drug primaquine caused haemolytic anaemia in some individuals. On further investigation, these individuals were found to have abnormally low levels of erythrocyte G6PD activity. (Australian Medical association, 1981)
Incidence
G6PD deficiency being X – linked has a higher incidence in males compared to females. This can be explained since men can only be hemizygous for the gene therefore their phenotype can only either be normal or G6PD deficient. Females have two copies of the gene therefore they stand a higher chance of being normal as one of the X chromosome is usually inactivated. (Motulsky & Yoshida, 1969)
Genetic and Molecular Basis of g6pd Deficiency
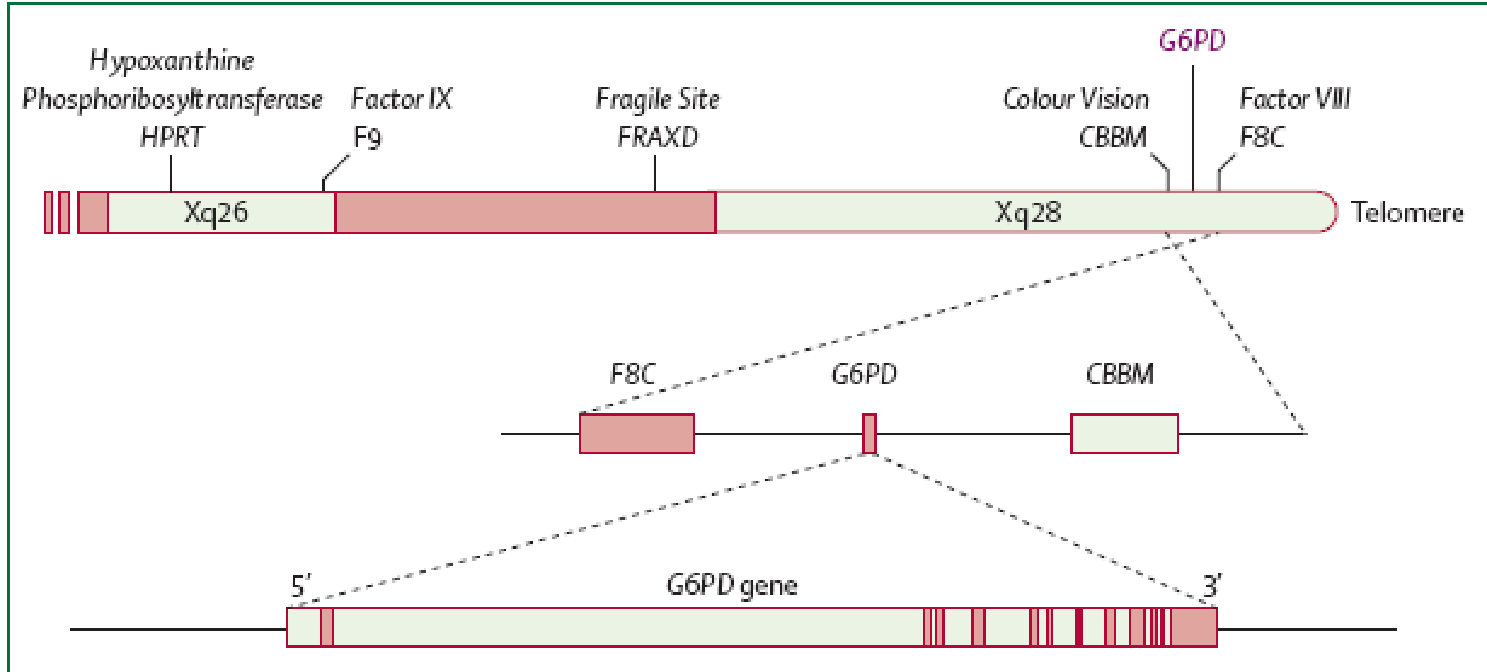
As stated earlier the main aetiology of glucose 6 phosphate deficiency is genetic with a typical X-linked pattern. Heterozygous females due to the normal X chromosome inactivation, tend to be genetic mosaics and therefore, they can have the same susceptibility as males to the same patho-physiological phenotype. They however suffer less severe clinical manifestations. The determinant G6PD gene is located on the long arm of X chromosome specifically at the telomeric region close to where the genes for colour blindness, haemophilia A and congenital dyskeratosis are located. After several DNA sequencing analysis of the various mutant forms, courtesy of the WHO, of G6PD the following features emerged:
- A significant number of the variants were attributed to single point mutations that had the resultant effect of amino acid substitution. So far only three deletions have been identified.
- Initially, the various variants were seen to be distinct however it emerged that they were identical based on their biochemical characteristics.
- Other than mutations polymorphism has been used to explain the presence of the different variants. Several polymorphic sites have been isolated in introns supporting the existence of G6PD haplotypes.
Classes of g6pd Deficiency
The disease is classified according to the level of enzymatic activity and its association with hemolytic anaemia. The classes include:
Class I
Severely deficient, associated with chronic non-spherocytic haemolytic anaemia. Here the residual enzymatic activity is below critical level thus NADPH production is insufficient for the red blood cell leading to chronic non spherocytic anaemia.
Class II
Severely deficient (1–10% residual activity), associated with haemolysis occurring more intermittently whereas the anaemia occurs only sporadically.
Class III
Moderately deficient (10–60% residual activity) and haemolysis occurs secondary to infections or other forms of stimulation.
Class IV
Normal activity (60–150%) Is largely non deficient with an above average enzymatic activity compared to those deficient of the enzyme.
Class V
Increased activity (>150%) (Central Society for Clinical Research, 1984)
Epidemiology
There is a worldwide distribution of deficient G6PD alleles with an approximate 400 million people as carriers. The areas with high prevalence of the same include southern Europe, Africa, southeast Asia, middle east, North and South America. It is interesting that the worldwide malaria distribution is almost the same as that of mutated alleles of G6PD confirming the notion that G6PD deficiency protects against malaria. (David, 1993)
Clinical Manifestations
According to (Golan, 2007), it is fortunate that a good number of G6PD-deficient persons do not suffer any related symptoms and are usually not aware of their state. Ideally, the disease presents with an acute case of haemolysis which occurs when the red blood cells are exposed to drugs, other infections like diabetes and myocardial infarction and at times physical stress that trigger oxidative reactions. The manifestations include:
- Infection-induced haemolytic anaemia.
- Chronic non-spherocytic haemolytic anaemia.
- Drug induce haemolysis.
- Favism.
- Neonatal jaundice.
Clinically the deficiency is characterized by symptoms like fatigue, anaemia, back pain splenomegaly and jaundice
Drug Induced Haemolytic Anaemia
Antimalarials, sulfonamides, sulfones, (Grossman, Budinsky & Jollow, 1995) nitrofurantoin, antipyretics e.g. aspirin and other chemicals like naphthalene are some of the drugs implicated in acute hemolysis in G6PD deficient individuals. It is not easy to determine whether a particular drug would precipitate a haemolytic crisis in G6PD deficient individuals. This is because one cannot extrapolate the effects of a drug on some patients to all other persons. This is solely because of the variability in pharmacokinetics among different individuals. Also, some drugs with a likelihood of oxidant effects are sometimes given to one who has an underlying clinical condition leading to haemolysis. (Beutler, 1978) Patients reactions may also vary because they are taking more than one medication at a time. Lastly in G6PD deficiency, there is tendency of the acute haemolysis to self resolve and therefore does not always produce lead to anaemia or reticulocytosis of clinical magnitude. There are several factors that enhance one’s susceptibility to, and extent of drug induced haemolysis and they can either be inherited or acquired. (Dacie, 1999) They include:
Inherited
- Actual nature of the defect in the enzyme.
- Metabolic integrity of red blood cell.
- Pharmacokinetic differences that are genetic in origin.
Acquired
- The age of the patient.
- Effect of the drug on enzymatic activity.
- Pre-existing concentration of haemoglobin.
- Presence of additional sources of oxidative challenge like an infection.
- The dosage and pharmacodynamics of the drug. (Ephinstone & Simon, 2009)
The haemolysis in drug induced haemolytic anaemia is detectable clinically as dark urine due to haemoglobinuria and jaundice arising after taking a certain drug. The anaemia tends to worsen and eventually recover after drug cessation. To detect this occurrence, methyl violet staining is used to detect the precipitates of denatured hemoglobin in the red blood cells of peripheral blood.
Infection- Induced Haemolytic Anaemia
This is a typical cause of hemolytic anaemia in people suffering from G6PD deficiency. This haemolytic state may be exacerbated by the concurrent administration of a particular drug, age of the patient or the liver function. Numerous bacterial, viral and rickettsial infections have been implicated as precipitants but of importance, infections like pneumonia, hepatitis A and B, typhoid and cytomegalovirus are notably implicated in causing haemolysis. In G6PD deficient children, viral infections of the upper respiratory tract or gastrointestinal tract cause more extensive haemolysis than bacterial infections. (Stuart et al, 2009) The infection induced haemolysis is largely intravascular with renal failure acting as a common complication in adults. The bilirubin levels specifically the total bilirubin levels could be elevated in a concomitant case of hepatitis together with a G6PD deficiency induced acute haemolysis. This can act as a likely source of errors in diagnosis when the haemolysis is a precipitant of the hepatitis. The clinical progression of a case of severe haemolysis may be significantly improved by giving prompt blood transfusions. If it occurs in acute renal failure it has a significant risk of complicating to tubular necrosis due to obstruction by haemoglobin casts. (Glader, 2009)
Favism
The ingestion of fava beans in G6PD deficient individuals led to the advent of the above named disorder as it was observed that these people developed a case of acute hemolytic anaemia. The haemolytic crisis as a result of fava bean ingestion was more severe than that caused by drugs or infection. (Gregg & Prichal, 2008) Based on studies by the American Federation of Clinical Research, the toxic components of the coveted Fava beans is attributable to the pyrimidine aglycones, divicine and isouramil in combination with ascorbic acid. These substances cause a disturbance in the erythrocyte homeostasis specifically by reducing the activity of the membrane calcium Atpase. This has the effect of increasing the intraerythrocytic calcium and a drop in the intracellular red blood cell potassium level. Beutler, 1993 suggests that this ionic disturbance mediates the activation of proteolytic activity within the red blood cell.
Neonatal Jaundice
It has been found that up to a third of male newborns with G6PD deficiency have neonatal jaundice. This may be attributed to cultural, genetic and environmental effects such as herbal remedies and oxidant drugs which the mother is exposed to. (Stuart et al).
Congenital Non-spherocytic Haemolytic Anaemia
This kind of anaemia is usually detected in early childhood (Scwartz, 2002) and most patients have suffered from neonatal jaundice, chronic anaemia worsened by oxidative stress and usually have G6PD deficiency in all tissues and rarely associated with granulocyte deficiency and dysfunction. It is a chronic form of haemolysis which is mainly extravascular. (Hoffmann, Zschoske & Nyhan, 2009)
In addition, acute or chronic forms of erythrocyte hemolysis that are attributable to G6PD deficiency are worsened by co inherited alterations in red blood cells such as defects in the membrane, thalassemia, pyruvate kinase deficiency and congenital dyserythropoietic anaemia. (Goldman & Ausiello, 2004)
Experiment
As briefly outlined above, glucose 6 phosphate dehydrogenase deficiency does cause haemolysis significant enough to lead to anaemia. The main objective of this paper was to back up this statement experimentally. To do this, we would have to simulate a g6pd deficient variant in the red blood cells by inhibiting the enzyme or minimizing its activity, thereafter subject it to oxidative stress as would normally occur and observe for the occurrence and degree of haemolysis.
Materials
- G6pd enzyme inhibitor preferably pyridoxal-5-phosphate.
- Magnesium chloride and triethanolamine as buffers.
- Trypan blue and haemocytometer to determine cell counts.
- Hydrogen peroxide as a source of oxidative stress.
- Sheep red blood cells.
Methodology
Pyridoxal 5’ phosphate acts by competitive and non competitive inhibition of glucose 6 phosphate substrate and NAD/NADP production respectively. (Scriver, 1995) You therefore begin by finding out what concentration of the inhibitor would be sufficient to derail the enzymatic activity in vitro. You can achieve this by carrying out an enzymatic assay of G6PD activity on cells that have been mixed with the inhibitor. (American Federation for Clinical Research, 1988)
Assay of G6PD activity
The enzyme activity was determined at 340 nm in a spectrophotometer. The reaction mixture contained 0.12 M Triethanolamine buffer (pH 7), 10 mM MgCl2 , 0.7 mM NADP , 2 mM glucose 6-phosphate and the appropriate amount of the enzyme solution. One unit of enzyme activity was taken as the number of µmole of NADP reduced per min at 30°C. (Beutler, 1984)
On finding the appropriate inhibitory concentration, you can use this or a higher value in vivo with sheep red blood cells to inhibit the enzyme and hence mimic a mutant form of G6PD with reduced or absent activity. Then count the number of haemolysed red blood cells and record your findings. (Beutler, 1993)
However, the use of inhibitor alone absent of any source of oxidative stress may not yield significant results as there is no actual stress on the cells. Therefore, a potential source of stress comes in handy to challenge the cells. You can use hydrogen peroxide by finding a concentration that just starts to kill a few cells, then use this together with the inhibitor to see if the inhibitor increases the proportion of dead cells. (Cammack & Attwood, 2006) You should also do the same for a separate group of cells without the inhibitor.
Thereafter, expose the cells to the appropriate conditions such as the right temperature and administration of glucose to stimulate the hexose monophosphate shunt for production of NADPH and reduced glutathione for both groups of cells. Then assess the cells using trypan blue staining and haemocytometer to determine the proportion of live/ dead cells. Record these results. You can additionally assess the nature and extent of damage to the cells by doing separate assays for methaemoglobin (product of haemolysis), TBARs and residual enzyme activity. (Bernadette , Fritsma & Kathryn, 2007)
Results
Immediately on addition of oxidative challenge, there was a 10 fold rise in the number of haemolysed red blood cells which was additionally supported by the significant production of products of red blood cell degradation i.e. methaemoglobin, TBARs and residual enzymes for the cells that had been inhibited. As for the uninhibited cells there was little or insignificant haemolysis. (Hohl, Kennedy & Frischer, 1991) The cells affected by the inhibitor were unable to restore the reduced glutathione levels upon stimulation of the hexose monophosphate shunt. The inhibitor acted to mimic a variant of mutated or absent G6PD hence ideally simulating its deficiency whereas the hydrogen peroxide served to simulate the various forms of oxidative stress listed above that cause the cell to be haemolysed. It is worth noting that there was also less haemolytic effect when the inhibitor was used on its own.
Discussion
From the above experiment it is sure that the glucose 6 phosphate dehydrogenase deficiency is a primary cause of haemolytic anaemia. The mechanism behind the haemolysis can be attributable to a reduced production of NADPH and subsequently reduced forms of glutathione. NADPH serves to donate electrons for the enzyme catalysed biosynthesis. The major contribution of the enzyme G6PD via production of the NADPH is that it confers reducing power to cells especially erythrocytes. (Mengel, Frei & Nachman, 1972) This occurs as the produced NADPH is used in reductive synthesis leading to production of reduced glutathione. The reduced glutathione on the other hand gives the cell the ability to counterbalance any form of oxidative stress from hydrogen peroxide and oxygen radicals, as well as maintaining haemoglobin and other erythrocyte proteins in the reduced state. (Hoffman, Benz & Shattil, 2005) A reduced glutathione level makes several sulphydryl groups in the proteins not to be maintained in the reduced form. Due to this, intra molecular and extra molecular disulphides are formed and such aggregates decrease red cell deformability eventually altering the cell surface sufficiently to make it easily recognizable by macrophages as being abnormal hence leading to a case of extravascular haemolysis within the reticulo-endothelial system.(Greg & Prichal, 2008)It is thus safe to say that haemolysis is the major pathophysiologic manifestation of the disease. The reason why some of these patients may be asymptomatic is because they may have the mutant form but do not get to be exposed to conditions that may challenge their cellular handling of oxidative stress. (Robert, Samuel & Thomas, 2003)
This experiment helps us come up with a management plan in handling the G6PD deficiency. First, it enables us come up with ways of definitive diagnosis of the disease. This should be based on enzyme activity estimation by a quantitative analysis of the production of NADPH from NADP. This can be done photometrically. In cases of rapid population screening, several semi quantitative tests can be applied such as dye-decolourisation and fluorescent spot tests. However, there can arise diagnostic issues when measuring the enzyme activity of various forms of the deficiency. The level of activity in young red blood cells is higher than in the mature erythrocytes and in a case of acute haemolysis in the presence of a high reticulocyte count a false negative value is likely. (Smith, 1979) There can also occur some difficulties in asssessing a neonate who has a significant amount of young red blood cells. For new variants of the disease, the World Health Organization recommends a complete biochemical characterization of the G6PD enzyme. Polymerase Chain Reaction tests, direct sequencing and denaturing gradient gel electrophoresis are some of the molecular methods that are employed in identifying specific mutations. Essentially, G6PD deficiency testing should be a prerequisite in cases of acute haemolytic reactions following exposure to a known oxidatively challenging drug, infection or fava bean ingestion in people from the areas where the condition is most prevalent.
It therefore goes without saying that the most effective way to tackle its debilitating effects is by preventing the haemolysis by essentially avoiding the cellular oxidative stressors e.g. drugs and fava beans. (Okpako, 1991) For this to be effective there has to be adequate patient awareness as to their condition, as a consequence of prior episodes of haemolysis or via a screening programme. Essentially, it should be one of the main differentials in cases of haemolytic anaemia. It is fortunate that the cases of acute haemolysis is usually short-lived and once the oxidative trigger is removed it resolves but some extreme cases require blood transfusion. The degree of anaemia in congenital non spherocytic haemolytic anaemia may worse if there are other exacerbating events and thus need monitoring. It is not clear whether antioxidants such as selenium and vitamin E (Vanella et al) have any effect but there is some evidence in case of chronic haemolysis whereas gene therapy is still under consideration. Splenomegaly and gallstones are the major complications of haemolysis.
Conclusion
Haemolytic anemia is a serious condition with G6PD deficiency being its major contributor. With as high as a worldwide prevalence of 400 million people acting as carriers of the G6PD deficiency gene, this disease should be accorded more concern. It is a good thing that most of these people will not have any clinical consequences. Unfortunately, for that proportion of affected individuals who develop a case of acute haemolytic anaemia or neonatal jaundice improper management could cost them their lives or permanent neurological damage. The high prevalence areas are coincidentally malaria-endemic areas such as tropical Africa and sub tropical Asia. These high prevalence areas should be on the lookout for the clinical manifestations of the disease especially clinicians and patients. To do this they have to institute the following:
- Where there is a high index of suspicion based on clinical and haematological findings, there should be an almost immediate follow up of quantitative spectrophotometric measurement of red blood cell activity. For large screening programmes, rapid fluorescent tests should be done confirmed by specific quantitative assay. (Crowther et al, 2008)
- On the patient’s side, avoidance is of what triggers the haemolytic events is key and also should be taught of the risks of the episodes and how to recognize them once they occur.
- For familial history of haemolysis, ethnic or geographic predisposition or neonatal jaundice should be tested in advance for G6PD deficiency early in advance.
List of References
American Federation for Clinical Research, 1998, Journal of investigative medicine: the official publication of the American Federation for Clinical Research, Volume 46, Issue 1, Slack, Northwestern University.
Australian Medical Association, British Medical Association, 1981, Medical journal of Australia, Volume 2, Issue 68, Australasian Medical Pub. Co., Pennsylvania State University.
Bernadette, F, Fritsma, G & Kathryn, D, 2007, Hematology: clinical principles and applications, 3rd edition, Elsevier Health Sciences, New York.
Beutler, E, 1978, Hemolytic anemia in disorders of red cell metabolism, Plenum Medical Book Co., California.
Beutler, E, 1984, ‘Red cell metabolism.’ In: A Manual of Biochemical Methods, 3rd edition, Grune and Stratton, New York.
Beutler, E, 1993, ‘The molecular biology of enzymes of erythrocyte metabolism.’ In: The Molecular Basis of Blood Disease, WB Saunders, Philadelphia.
Cammack, R & Attwood, T, 2006, Oxford dictionary of biochemistry and molecular biology, 2nd edition, Oxford University Press, Oxford.
Central Society for Clinical Research (U.S.) 1984, The Journal of laboratory and clinical medicine, Volume 104, C. V. Mosby, Michigan.
Crowther , M, Ginsberg , J, Meyer, R, Schünemann, H & Lottenberg, R, 2008, Evidence-Based Hematology, Volume 21 of Evidence-Based Medicine, John Wiley and Sons, New York.
Dacie , J, 1999, The haemolytic anemias: Drug- and chemical-induced haemolytic anaemias, paroxysmal nocturnal haemoglobinuria, haemolytic disease of the newborn, 3rd edition, Churchill Livingstone, Chicago.
David, A, 1993, Genetic factors in drug therapy: clinical and molecular pharmacogenetics, Cambridge University Press, Cambridge.
Elphinstone, J & Simon, S, 2009, General and systematic pathology, 5th edition, Churchill-Livingston, New York.
Glader, B, 2009, ‘Hereditary hemolytic anemias due to red blood cell enzyme disorders’ Wintrobe’s Clinical Hematology, 12th ed, p.933, Lippincott Williams & Wilkins, Philadelphia.
Glew, R, Rosenthal , R & Rosenthall, D, 2009, Medical biochemistry: human metabolism in health and disease, John Wiley and Sons, New York.
Golan, D, 2007, ‘Hemolytic anemias: red cell membrane and metabolic defects.’ Cecil Medicine, 23rd ed., chapter 165, Saunders Elsevier, Philadelphia.
Gregg, X & Prichal, J, 2008, ‘Red blood cell enzymopathies,’ Hematology: Basic Principles and Practice. 5th ed. , chap 45, Churchill Livingston, Philadelphia.
Grossman, S, Budinsky, R & Jollow, D, 1995, ‘Dapsone-induced hemolytic anemia: role of glucose-6-phosphate dehydrogenase in the hemolytic response of rat erythrocytes to N-hydroxydapsone.’ Journal Pharmacol Exp Ther, 273(2), 70-7.
Hoffmann , G, Zschocke, J & Nyhan, J, 2009, Inherited Metabolic Diseases: A Clinical Approach, Springer, Los Angeles.
Hoffman, R, Benz, E & Shattil, J, (eds.) ,2005, Hematology: Basic Principles and Practice. 4th ed., Churchill Livingston, Philadelphia.
Hohl ,J, Kennedy ,J & Frischer, H, 1991, Defenses against oxidation in human erythrocytes: role of glutathione reductase in the activation of glucose decarboxylation by hemolytic drugs. Journal of Lab Clin Med, 117(4), 325-31.
Mengel , C, Frei, E & Nachman, R, 1972, Hematology; principles and practice, Medical Publishers, New York.
Motulsky, A & Yoshida, A, 1969, ‘Methods for the study of red cell, glucose-6-phosphate dehydrogenase.’ In: Biochemical Methods in Red Cell Genetics, Academic Press, New York.
Murray, R, Granner, D, Mayes, P, & Rodwell, V, 2003, Harper’s Illustrated Biochemistry, 26th edition, McGraw-Hill Medical, Michigan.
Okpako, D, 1991, Principles of pharmacology: a tropical approach, Cambridge University Press, Cambridge.
Robert, I , Samuel, E & Thomas, P, 2003, Blood: principles and practice of hematology, 2nd edition, Lippincott Williams & Wilkins, Newyork.
Schwartz, M, 2002, Five-minute pediatric consult ‘The 5-Minute Consult Series’, 3rd edition, Lippincott Williams & Wilkins, Philadelphia.
Scriver, R, 1995, The metabolic and molecular bases of inherited disease, 7th edition ,Volume 3, McGraw-Hill, Health Professions Division, Minnesota.
Smith , G, 1979, The Yale journal of biology and medicine, Volume 52, Yale Journal of Biology and medicine, Yale.
Stuart, H, Nathan, D, Ginsburg, D & Thomas ,A, 2009, Nathan and Oski’s hematology of infancy and childhood, 7th edition, Elsevier Health Sciences, Chicago.
Vanella, A, Campisi, A, Castorina, C, Sorrenti ,V, Attaguile, G, Samperi ,P, Azzia, N, Di Giacomo, C & Schiliro G, 1991, Antioxidant enzymatic systems and oxidative stress in erythrocytes with G6PD deficiency: effect of deferoxamine, 24 (1),25-31.