Introduction
In the past decade, there have been important advances made in the treatment of various forms of cancer (Demo, Krik, & Aujay 2007). The discoveries made so far have led to increased survival rates for individuals affected with considerably risky forms of cancer. The introduction of drugs like thalidomide and the discovery of other novel treatments such as proteasome inhibitors has enhanced the fight against multiple myeloma (MM) (Meng, Mohan, Kwok, Elfsson, and Crews 1999). The major challenge has been preventing MM from relapsing or becoming refractory to treatment (Tanday 2012). According to the Multiple Myeloma working group, the novel agents have shown poor results in the treatment of relapsed and refractory multiple myeloma. However, the hope of finding a solution has increased with the discovery of carfilzomib. Carfilzomib is a next-generation, selective proteasome inhibitor that is undergoing evaluation for use in the treatment of refractory and relapsed multiple myeloma (Andrzej, et al. 2012). Ongoing clinical trials have shown that carfilzomib has a positive efficacy with a good safety profile (Meng, Mohan, Kwok, Elfsson, and Crews 1999). The drug is administered intravenously for 2-10 minutes, twice a week for about 3 weeks, followed by a 12-day rest period in a treatment cycle that lasts for 28 days (Onyx Pharmaceuticals 2012). This paper reports on the following: carfilzomib drug profile; detailed review of multiple myeloma; drug design; mechanism of action and the use of the drug for treatment.
Multiple myeloma (MM)
Multiple myeloma can also be identified as plasma cell myeloma or Kahler’s disease (Demo, Krik, & Aujay 2007). Myeloma can be described as cancer that affects plasma cells. The accumulation of abnormal white blood cells in the bone marrow due to the condition is then referred to as multiple myeloma. According to Tanday (2012), “the accumulation of the abnormal cells in the bone marrow interferes with the production of normal white blood cells.”
Multiple myeloma can affect several organs in the body and, therefore, symptoms can vary significantly. In certain instances, a mnemonic is often used to remember the four parts of multiple myeloma (Tanday 2012). The mnemonic (CRAB) identifies the major indications of MM, which include the following: C identifies the elevated calcium levels that are seen in MM, R refers to renal failure, A denotes anemia, while B refers to bone lesions that are often observed in multiple myeloma. The disease can have many different symptoms, some of which may be the result of other causes. Common symptoms are described below.
Bone pain
Close to 70% of individuals suffering from multiple myeloma experience bone pain. Most of the time, it is possible for the ribs and spine to experience constant pain, sometimes, it is increasing. Also, according to Onyx Pharmaceuticals (2012), “a pathological bone fracture may result into a persistent localized pain.” Myelomas that result in bone disease are usually a result of overexpression of Receptor Activator for Nuclear Factor kB ligand (RANKL) by the bone marrow stroma (Onyx Pharmaceuticals 2012). The lesions that develop due to the condition cause breakdown of the bone structure. This in turn results in elevated blood calcium levels (Onyx Pharmaceuticals 2012).
Infection
Pneumonia and pyelonephritis are other widespread infections. Infections occur due to immune deficiency and the onset is usually observed following chemotherapy (Kane, Bross, Farrel, and Pazdur 2003). The clonal plasma cells produce ineffective clonal antibodies. The risk of infection is usually reduced using replacement immunoglobulin therapy.
Renal failure
According to Tanday (2012), “both acute and chronic renal failure can develop in individuals suffering from multiple myeloma.” Renal failure usually occurs due to elevated levels of calcium. It can also occur due to the excretion of light chains (Bence Jones proteins). Light chains are manifested as the Fanconi syndrome (Tanday 2012).
Anemia
“Both normocytic and normochromic anemia can be found in association with myeloma” (Tanday 2012). Anemia progresses when the bone, being in the normal state, is changed for the infiltrating tumor cells which have the production of normal white blood cells through cytokine action.
Neurological manifestations
There are several neurological problems that are observed in multiple myeloma conditions. The most common conditions include weakness, fatigue, and confusion, and occur due to elevated levels of calcium. Patients may also develop retinopathy and headaches and this is due to blood hyperviscosity that is dependent upon the properties of paraprotein (Meng, Mohan, Kwok, Elfsson, and Crews 1999). Other neuropathies such as the loss of bladder and bowel control may occur due to increased involvement of the spinal cord (Demo, Krik, and Aujay 2007).
Diagnosis
Diagnosing of MM is prompted following the “establishment of kidney dysfunction, unexplained anemia, high erythrocyte sedimentation rate (ESR), elevated levels of beta-2 microglobulin, and high serum protein” (Onyx Pharmaceuticals 2012). Following the establishment of these conditions, a workup to investigate MM is initiated. The workup basically involves a skeletal survey using techniques such as x-ray, magnetic resonance imaging (MRI), and CT scan to visualize bone lesions.
Bone marrow biopsy is carried out to establish the fraction of plasma cells in the bone marrow. Immunochemistry is used to flag the defective plasma cells.
Commercial immunoassay techniques that detect free light chains are currently available and are useful in the identification of disease progression and suitable treatment strategies.
Treatment
Historically, the treatment of MM has been focused on strategies that decrease the population of clonal plasma cells. This in turn reduces the harmful symptoms of the disease. Other treatment strategies are also used to minimize the likelihood of bone fractures. The treatment of multiple myeloma is complex and depends on several factors such as the age of the patient and comorbidities (Siegel, et al. 2012).
Stem cell transplantation is also indicated in severe cases. Induction chemotherapy is carried out prior to the transplantation. Common induction regimens include “thalidomide-dexamethasone, bortezomib regimens, and lenalidomide- dexamethasone” (Kane, Bross, Farrel, and Pazdur 2003).
However, in patients whose transplantation cannot be done for one reason or another, chemotherapy is often given as the standard disease management strategy. Treatments with drugs such as bortezomib and prednisone have been shown to significantly increase survival rates. Research into the development of new drugs is ongoing and this is where Carfilzomib comes in.
Relapsed and refractory multiple myeloma
The development of therapeutic regimens that include bortezomib and a number of IMiDs has been instrumental in the improvement of outcomes for patients with new cases of MM (Onyx Pharmaceuticals 2012). However, MM still remains incurable and eventually fatal, even in patients who show good responses in the initial treatment stage. The duration of survival time was improved by the development of SCT in the 1990s, with improvements being made in the 2000s. This coincided with the discovery of more novel agents such as thalidomide, lenalidomide, and bortezomib (Onyx Pharmaceuticals 2012). These novel drugs are usually used alone or in combinations to tackle recurrent cases of MM. Resistances usually emerge when patients are repeatedly treated with these agents over a long duration. The use of novel agents in the treatment of MM also results in cumulative toxicities which in turn contribute to the development of comorbid conditions (Demo, Krik, & Aujay 2007).
The lack of a standard regimen for the treatment of patients with relapsed and refractory myeloma has made a strong case for the development of Carfilzomib. The International Myeloma Working Group (IMWG) has made efforts to describe the nature of relapsed and refractory MM, and the poor results are seen when novel agents are used to treat such cases.
Drug Design
Carfilzomib is a drug derived from epoxomicin, a natural proteasome inhibitor (Siegel, et al. 2012). Research on epoxomicin was pioneered by Craig Crews laboratory at Yale University. Further studies conducted by the Crew led to the invention of a more specific derivative known as YU101 (Andrzej, et al. 2012). The derivative was licensed to ProteolixInc, who in turn modified it to develop Carfilzomib (Onyx Pharmaceuticals 2012). Carfilzomib has been subjected to a number of clinical trials that include Phase 1 and Phase two trials, and a phase two pivotal trial that is intended to achieve a faster approval. The drug has been found to provide meaningful clinical benefits to patients with relapsed and refractory cases of MM (Onyx Pharmaceuticals 2012).
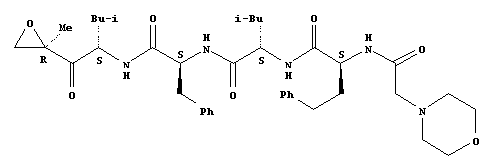
Synthesis or Extraction
The chemical formula of Carfilzomib is C40H57N5O7. The drug is isolated in crystalline-free base form (Siegel, et al. 2012). The multi catalytic proteinase complex nature of the drug enables it to degrade a large number of protein substrates being found in normal and transformed cells (Siegel, et al. 2012). The drug is described as a tetrapeptideepoxyketone that selectively and irreversibly binds to cause the inhibition of the 20S proteasome, thereby resulting in the apoptosis of cancer cells (Kane, Bross, Farrel, & Pazdur 2003). The proteasome inhibition induced by Carfilzomib is “more durable and potent compared to bortezomib” (Siegel, et al. 2012).
Provide at least one route to obtaining pure drug material, e.g. total or partial synthesis, fermentation & extraction as appropriate.
Biochemical Mode of Action
Carfilzomib targets intracellular proteins in the abnormal plasma cells. The targeted intracellular proteins are first “ubiquitinated through the ubiquitin conjugation system and then cleaved within the 20S core of the proteasome” (Andrzej, et al. 2012). This action is facilitated through the following enzymatic activities: “chymotrypsin-like (CT-L) activity, trypsin-like activity, and the caspase-like activity” (Meng, Mohan, Kwok, Elfsson, & Crews 1999). Subunits found in the proteasome active sites are composed of unique proteases that have the “N-terminal threonine as the active site nucleophile” (Siegel, et al. 2012). Carfilzomib is classified as a second-generation proteasome inhibitor and results in the formation of an irreversible double covalent adduct with the N-terminal threonine residue of the proteasome active sites (Myung, Kim, Lindsten, Dantuma, & Crews 2001). A more potent covalent bond is formed when the active site is a CT-L active site of the constitutive proteasome (Beta5 subunit) and immunoproteasome (Onyx Pharmaceuticals 2012). Therefore, the proteasome inhibition that occurs when Carfilzomib is used is more potent, specific, and durable when compared to bortezomib (Siegel, et al. 2012).
In tumor cells, proteasome inhibition leads to the accumulation of ubiquitinated proteins, proteotoxic stress, and, finally, apoptosis (Andrzej, et al. 2012). Research studies have shown that the CT-L mediated inhibition accomplished via the Beta5 and LMP7 is sufficient enough to induce cytotoxicity (Andrzej, et al. 2012). Studies conducted on Carfilzomib have shown that the drug can inhibit CT-L proteasomal activity in purified preparations of human 20S enzyme (Onyx Pharmaceuticals 2012). Incubation of hematologic tumor cell line with Carfilzomib leads to high rates of proteasomal inhibition that is followed by the accumulation of polyubiquitinated proteins and the induction of apoptosis and eventual cell death (Tanday 2012). In a study conducted in 2007, it was shown that Carfilzomib was able to induce high potent apoptosis in cells derived from patients suffering from relapsed and refractory MM and have been to be resistant to bortezomib.
A study conducted by Bennett in 2008 established that carfilzomib had high selectivity for proteasomal N-terminal as opposed to bortezomib which inhibits a number of proteases including “cathepsins A, and G, dipeptidyl peptidases II, chymase, and HtrA2/omi” (Siegel, et al. 2012). The potency exhibited by Carfilzomib is comparable to that of bortezomib both in vivo and invitro. Investigations conducted using neuronal cells showed both bortezomib and carfilzomib had similar protein inhibition potencies, however, only bortezomib was able to result in the degeneration and eventual death of neuronal cells. The difference was observed by the fact that bortezomib and not carfilzomib was able to inhibit the action of HtrA2/omi, a protease that has excellent survival activity in neuronal cells.
The inability for carfilzomib to result in the death of neuronal cells can be explained by the lack of neurobehavioral or peripheral nerve histologic in animal models that have been subjected to prolonged carfilzomib treatment.
Maximum tolerant administration of bortezomib and carfilzomib in rats showed that carfilzomib had higher proteasome inhibition levels in tissues and bone marrow (Meng, Mohan, Kwok, Elfsson, and Crews 1999). This indicates that the ability to tolerate higher doses of carfilzomib results in better proteasome inhibition. The administration of carfilzomib to rats in a QDx2 weekly dose schedule showed a significant reduction in tumor development, particularly for mice that had human colorectal adenocarcinoma. This finding suggests that dose-intensive schedules can prevent tumor growth through prolonged proteasome inhibition.
Prodrugs: rationale; uptake, distribution site, and mechanism of activation
Carfilzomib is a proteasome-based drug. The proteasome is one of the two main pathways of protein degradation, the other being the lysosome pathway. Whereas lysosomes degrade extracellular proteins, proteasomes mainly work on intracellular proteins (Demo, Krik, and Aujay 2007). As explained in the previous section, proteins are tagged for degradation by Carfilzomib by the ubiquitin tag (Onyx Pharmaceuticals 2012).
Proteasome inhibitors can be either synthetic or natural and currently exist in five classes which include peptide aldehydes, peptide boronates, peptide epoxiketones and beta-lactones, and peptide vinyl sulphones. Carfilzomib belongs to the epoxiketones class of proteasome inhibitors.
Carfilzomib is classified as a “multicatalytic proteinase complex that degrades a wide variety of protein substrates in normal and transformed cells” (Myung, Kim, Lindsten, Dantuma, and Crews 2001).
Carfilzomib (Kyprolis) has been approved for intravenous injection in the United States. The drug is indicated for treatment in patients with MM and who have gone through at least two other therapies that may include bortezomib and an immunomodulatory agent (Demo, Krik, & Aujay 2007). The drug is administered intravenously for a duration of between 2 and 10 minutes for two successive days every week for a total of three weeks. This is then followed by resting for a total of 12 days. The recommended cycle is 20mg/m2/day and can be increased if tolerated (Tanday 2012). Patients who are about to administer the drug should be hydrated to mitigate the risk of tumor lysis syndrome and renal toxicity.
Following administration, Carfilzomib achieves a steady volume distribution of 28L. In vitro testing shows that the drug achieves an average of 97% binding to human plasma proteins at a concentration that ranges from 0.4 -4 uM (Onyx Pharmaceuticals 2012).
The drug undergoes extensive and rapid metabolization and releases peptide fragments and the diol in plasma and urine. Peptidase cleavage and epoxide hydrolysis are the principal metabolic pathways that Carfilzomib undergoes.
Following administration, the drug is rapidly cleared from the system with a less than one-hour half-life. The drug achieves a systemic clearance of between 151 and 263 L/hour.
Metabolism and Toxicology/uptake / PK/PD toxicology
Pharmacokinetics (PK)
Carfilzomib can be administrated via different routes. Studies have shown that when the drug is administrated intravenously at 15mg/m2 and 20mg/m2 in patients suffering from multiple myeloma or other solid tumors, the mean Cmax after the first dose is 2546/ng/mL and 2390-3060 ng/ml, respectively (Siegel, et al. 2012). When the drug was administered twice a week on consecutive days, then the Cmax values were found to be “1768ng/ml at 15mg/m2, 4026 at 20 mg/m, and 4232ng/ml at 27mg/m2” (Onyx Pharmaceuticals 2012). It was established that carfilzomib had a 28L mean volume of distribution at a steady state after the administration of 20mg/m2 of the drug. The drug bound human plasma proteins at approximately 97% at concentration levels that ranged from 0.4-4um.
The mean clearance values registered by carfilzomib(150-263 L/hr at 20 and 27 mg/m2) from the plasma compartment are higher than liver blood flow. This suggests that the drug is mainly cleared in an extra-hepatic environment. The drug undergoes a rapid and extensive systematic metabolic process. Carfilzomib generates a number of metabolites in urine, plasma, and in vitro hepatic cells. The metabolites include peptide fragments generated from the parent drug (M14 and M15) and the carfilzomibdiol (M16) (Siegel, et al., 2012). This indicates that cleavages by peptidase and epoxide hydrolysis are major pathways for carfilzomib metabolism. The metabolites are generated shortly after drug administration and they do not have epoxyketomepharmacophore (Onyx Pharmaceuticals 2012). The metabolites do not significantly inhibit proteasome. Unlike in other metabolites, the M15 shows a slight increase in total exposure that is related to the extent of renal impairment.
Carfilzomib is eliminated as inactive peptide fragments that are excreted in the urine. Less than 1% of the drug is excreted while still in the form of the parent drug. There is no data on the PK values in patients that have hepatic disorders.
Five studies were conducted to establish the variation of PK values from a total of 1488 samples from 236 patients (Demo, Krik, and Aujay 2007). The studies did not detect any effect of carfilzomib on the sex, age, or race of patients who were included in the study. The central compartment clearance showed some variation in creatinine clearance values (Siegel, et al. 2012). During the phase II 005 study conducted in patients with refractory and relapsed multiple myeloma and various levels of renal function, no statistically significant difference was observed in the clearance of carfilzomib.
Pharmacodynamics (PD)
In spite of the fact that carfilzomib clears rapidly in the plasma compartment, studies have shown that IV administration results in a more rapid proteasome inhibition that is observed in the whole blood and peripheral mononuclear cells (PBMCs) in patients that have MM or other solid tumors (Andrzej, et al. 2012). In the studies, doses that are below 15mg/m2 showed a consistent induction of 80% or more inhibition of the CTL-L proteasome activity in whole blood and PBMCs (LMP7 subunit of the immunoproteasome). The LMP2 and the MECL1 registered between 30%-40% and 40% -60% inhibitions of the immunoproteasome subunits, respectively (Andrzej, et al. 2012). The Beta1 and Beta2 subunits of constitutive proteasome were not inhibited by carfilzomib.
The over 80% rates of inhibition achieved in less than 48 hours that are registered when carfilzomib is administered to patients at doses that are less than 15mg/m2 are far much effective compared to corresponding values that are seen in bortezomib (Siegel, et al. 2012). The irreversible inhibition mechanism is responsible for the prolonged inhibition periods that are attained with carfilzomib. No differences have been observed between the degree of inhibition of the CT-L activity in whole blood samples and PBMCs when applied at a constant dose range varying between 15 and 27mg/m2. However, “the LMP2 and MECL1 subunits of immunoproteasome” showed an increasing inhibition trend with increasing dose (Myung, Kim, Lindsten, Dantuma, & Crews 2001). The levels of potent inhibition observed in the “CT-L activity and the tumor cells measured on the second day, and obtained from the patient’s bone marrow were comparable to measurements” taken from blood and PBMCs in other trials (Siegel, et al., 2012). This indicates that carfilzomib is evenly distributed in the blood following IV administration and the inhibitory activity that takes in the blood compares to the target inhibition that occurs in tumor cells. The whole recovery of proteasome activity was realized in the cycles between PBMCs and there was no change in proteasome content noted in the same cells (Onyx Pharmaceuticals 2012).
Toxicology
Administration of Carfilzomib in rats and monkeys within the safety range has been shown to “yield up to 80% inhibition of proteasome present in blood in a dose-intensive schedule” (Andrzej, et al. 2012). Such high rates of inhibition within a safety margin have not been achieved with bortezomib. The toxicology studies conducted in monkeys and rats complied with Good Laboratory Practice (GLP) and used employed two varied dose-intensive schedules with Carfilzomib (Siegel, et al. 2012). The dose-limiting toxicities observed were mainly related to the gastrointestinal and cardiovascular systems. This observation was made in both the 28-day cycle weekly schedule and the 28-day cycle two weeks cycle and in both species. There were no differences observed in subsequent repeat studies.
A number of nonclinical toxicities are associated with Cmax and are observed when using Carfilzomib. These include the increase in blood urea nitrogen and creatinine and studies have shown that the toxicities are often due to prerenal azotemia (Demo, Krik, & Aujay 2007). Other toxicity instances such as acute phase response and thrombocytopenia may occur due to proteasome inhibition and are varied. No hepatic toxicity is observed when the drug is administered acutely and chronically and this is most likely due to the extrahepatic clearance that of Carfilzomib.
Chronic dosing of the drug in animal models (9 months in monkeys and 6 months in rats) has not produced any significant neurobehavioral variations. In addition, the peripheral nerve sections have not shown any histologic changes following investigations conducted by neuropathologists (Andrzej, et al. 2012). These findings are very different and contrasting to those that were established when bortezomib was subjected to similar studies.
Principal uses/diseases: Opportunities and reasons for therapeutic intervention, choices of drugs, the rationale for the use
According to the information revealed in the sections above, Carfilzomib is a next-generation proteasome inhibitor that could play a leading role in the treatment of patients diagnosed with relapsed and refractory multiple myeloma (Demo, Krik, and Aujay 2007). The open-label, single-arm study conducted in the second phase of drug investigation studies showed positive results.
The drug was approved for the treatment of multiple myeloma by the US food and Drug Administration (FDA) in July 2012 (Demo, Krik, and Aujay 2007). The decision to approve Carfilzomib was done quickly because there are no effective drugs that can be used to treat the advanced stage of MM.
Multiple myeloma patients receive cycles of Carfilzomib treatment that last for the duration of 3 to 4 weeks. For optimal results, the drug is administered intravenously two times a week. Treatment shows higher response rates in patients that have a less advanced MM. This finding is consistent with the potent proteasome inhibition properties of Carfilzomib (Onyx Pharmaceuticals 2012). However, previous studies have shown that this is not always the case and, therefore, data that is not randomized and non-concurrent requires caution while interpreting.
Combinations and Contraindications
Combinations
Due to the fact that bortezomib had shown poor results in regard to proteasome inhibition, particularly in patients suffering from relapsed and refractory MM, and therefore, there was a need for a more effective proteasome inhibitor. The development of the second-generation proteasome inhibitor (Carfilzomib) was a step in the right direction.
Identification of a combined drug regimen with the ability to increase the potency of Carfilzomib will be a good move. Though currently not undertaken, the combination of Carfilzomib and vorinostat may produce a potential synergistic interaction (Onyx Pharmaceuticals 2012). Research evidence shows that HDAC inhibitors activate the NF-kB survival pathway, while proteasome inhibition closes the pathway (Andrzej, et al. 2012). Available evidence also shows that proteasome inhibitors like bortezomib result in the formation of an aggresome that might promote cell survival. Such activity is, however, blocked by HDAC inhibitors. Additionally, the induction of ROS and oxidative stress generation has been shown in both proteasome and HDACI inhibitors (Demo, Krik, and Aujay 2007). Therefore, if the two drugs are combined then it is possible that a potent synergistic inter-action will take place. Various studies have proved the existence of a synergistic interaction between vorinostat and bortezomib in several different cancer types (Kane, Bross, Farrel, and Pazdur 2003). The different cancer situations include solid tumors, hematologic malignancies, and other clinical trials (Demo, Krik, and Aujay 2007). In light of this revelation, it has been proposed that Carfilzomib be combined with vorinostat in order to achieve a synergistic interaction that will promote cell death in both ABC DLBCL and GC cell lines (Meng, Mohan, Kwok, Elfsson, and Crews 1999).
Another independent study has established that the combination of Carfilzomib with dexamethasone at a low dose, and lenalidomide produces high activity in patients that have been newly diagnosed with multiple myeloma. The findings that make this observation was presented at the ‘2012 Annual Meeting of the American Society of Clinical Oncology” (Kane, Bross, Farrel, and Pazdur 2003).
Critical Appraisal
The development of Carfilzomib has generated new hope in the management and treatment of relapsed cases of MM. The epoxomicin-derived drug can be described as a natural protease inhibitor that results in irreversible protease inhibition. The irreversible inhibition has enabled the drug to manage cases of MM that were proving to be too challenging for existing drugs.
The use of Carfilzomib for the treatment of MM, particularly in the refractory and relapsed state has been approved by the FDA, though it is still undergoing studies intensive drugs. So far, the drug provides the best for individuals suffering from multiple myeloma. The drug’s ability to initiate an irreversible proteasome inhibition and the fact that it is cleared in the extra-hepatic pathway makes it a viable option for long-term use in MM patients.
However, the drug has some weaknesses that include the following: the drug produces a number of adverse events (AEs), “common ones being fatigue, anemia, nausea, thrombocytopenia, dyspnea, diarrhea, pyrexia, upper respiratory tract infection, headache, and increased serum creatinine” (Andrzej, et al. 2012).
Some serious adverse events (SAEs) have also been associated with the use of Carfilzomib. Those that were most commonly reported during trial studies include pneumonia, disease progression, and acute renal failure (Andrzej, et al. 2012). Renal failure and pneumonia cases were most commonly observed in patients with advanced disease states. Most of the deaths that were reported during trial studies were mainly linked with disease progression. However, in 2 out of the 14 patients who died, adverse events were identified as secondary causes of death (Andrzej, et al. 2012).
References
Andrzej, J., Dytfeld, D., Griffith, K., Lebovic, D., Vesole, D., Jagannath, S., et al 2012. ‘A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma’,bloodjournal.hematologylibrary.org, no.120, pp. 1801-1809.
Demo, S. D., Krik, C. J., & Aujay, M 2007. ‘Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome’,Cancer Res, no.13, pp. 6383-6391.
Kane, R. C., Bross, P. F., Farrel, A., &Pazdur, R 2003. U.S. ‘FDAapproval for the treatment of,’ Oncologist, no. 6, pp. 508-513.
Meng, L., Mohan, R., Kwok, B., Elfsson, M., & Crews, N. 1999. ‘Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity’,Proc Natl Acad Sci USA , no.18 pp. 10403–8.
Myung, J., Kim, K., lindsten, K., Dantuma, N. P., & Crews, C. 2001. ‘Lack of proteasome active site allostery as revealed by subunit-specific inhibitors’ , J mol, pp. 411–20).
Onyx Pharmaceuticals 2012. Proposed T rade Name: Kyprolis (Carfilzomib for Injection): Briefing Document. San Francisco: Onyx Pharmaceuticals, Inc.
Siegel, D., Martin, T., Wang, M., Vij, R., jakubowiak, A., Lonal, S., et al. 2012. ‘A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma’, bloodjournal.hematologylibrary.org, no. 120, pp. 2817-28255.
Tanday, S 2012. ‘Carfilzomib for multiple myeloma’ viewed October 30, 2012, www.thelancet.com/oncology, pp.373.