Introduction
The regulation of medical devices is considered to consist of some of the most important legislation written by governments. This is clearly due to the number of disciplines involved in the production of a single medical device. Medical device regulations affect both the safety of their users and the feelings of those dispirited people grasping at any hope to solve or improve a medical condition. The regulation of medical devices is a vast and swiftly developing field that is constantly complicated by governments’ legal technicalities.
With respect to devices to be sold, governments can make and enforce regulations to declare any class adulterated. Governments also monitor the labeling and packaging of the devices, as well as the offering, exposing and advertising for their sale. They also consider the size, dimensions, fullness and other specifications of packages involved in the manufacturing of medical devices. In addition, the regulatory agency is responsible for the regulation and conditions of the sales. Furthermore, regulatory agencies oversee the acceptable use of all materials as ingredients in order to prevent the purchaser or consumer from being deceived or misled in terms of design, construction, performance, intended use, quantity, character, value, composition, merit or safety, and to prevent any damage to the public’s health.
In this minor thesis, there will be a discussion about different elements that are currently under consideration by major regulatory agencies, such as the Therapeutic Goods Administration (TGA) in Australia, the Food and Drug Administration (FDA) in the United States and the European Union (EU) in Europe, that are relevant to the release of some important medical devices that apply energy to the human brain. The medical devices that will be covered are involved in deep brain stimulation (DBS), transcranial magnetic stimulation (TMS) and, finally, electroconvulsive therapy (ECT. In addition, an answer will be provided as to the sufficiency of the current regulations in place for the handling of such sensitive devices before their release onto the market and into public use.
Aims and Objectives
This minor thesis aims to study the impact of regulations that apply to the medical devices providing energy to brain on the safety and effectiveness of such devices. This aim of the thesis will be achieved by accomplishing the following objectives evolved in connection with the study of the regulations concerning safety of the medical devices.
- To study the background and application of medical devices regulation including the role of the United States Food and Drug Administration
- To Study the general aspects of medical device safety and risk management with respect to medical devices in general including the classification of the medical devices based on the risks associated
- To study the background and application of certain critical medical devices which apply energy to the human body.
- To study the issues relating to the issue of medical devices to public use
Significance of the Thesis
With the increase in the types and numbers of medical devices introduced in the market and improvements in the technology in manufacturing the devices has increased the chances of potential harm to the patients or users of these devices due to any default or manufacturing defects in the devices. Although FDA takes considerable precaution before according its approval for the release of any medical device for public use, it is practically impossible for the authority to control the safety and effectiveness of the devices. Therefore the study of regulatory measures controlling the safety of the devices at the design stage as well as in the post-market stage assumes greater significance to add to the existing knowledge on the subject. Especially in the case of those devices which provide additional energy to human brain the probability of the devices inflicting more harm to human beings due to malfunctioning or any fault in the device is more. For this reason it becomes necessary that there are stricter regulations monitoring the performance of these specialized and critical devices. This minor thesis through the study of regulatory measures monitoring the performance of the devices applying energy to human brains is expected to throw further light on the study of safety of medical devices.
The thesis is organized to have different chapters dealing with the different aspects of the study. Following this introductory chapter, chapter two contains the description of the background on medical devices regulation to expand the knowledge on the subject of study. Chapter three presents the salient aspects of certain critical medical devices that will be covered are involved in deep brain stimulation (DBS), transcranial magnetic stimulation (TMS) and, finally, electroconvulsive therapy (ECT). Chapter four discusses the role of regulatory authorities and the rules and regulations framed to ensure safety of the medical devices in general and the rules concerning the devices that apply energy to human brain in particular. Chapter five contains some concluding remarks in addition to providing few recommendations emerging out of the study under this minor thesis.
Background on Medical Devices Regulation
Standards
According to the International Organization for Standardization (ISO):
“Standards are documented agreements containing technical specifications or other precise
criteria to be used consistently as rules, guidelines or definitions of characteristics to
ensure that materials, products, processes and services are fit for their purpose.” [3]
Many types of specifications are present in standards. For example, there are prescriptive specifications that dictate product characteristics such as device dimensions and testing procedures. There are also design specifications that deal with such technical characteristics as operating room facilities and medical gas systems. Other types include performance specifications and management specifications. [4]
Recently, a new type of specification in standards has been developed, which is known as generic management system standards. As examples of this new specification, the ISO 9000 series consists of standards for quality management while the ISO 14000 series addresses environmental management. ISO 13485 and ISO 13488 are the generic management system standards specifically dedicated to medical devices. [5]
Standards exist for numerous reasons and purposes. Not only do they provide reference criteria for products to meet, they also provide any information that helps in enhancing the products’ safety, reliability and performance. In addition, standards give consumers in the marketplace increased confidence regarding products’ characteristics. [4]
Four familiar methods exist to assess a product’s conformity to a standard. The first is direct product testing. The second involves the audit of the manufacturing process and authorizing the manufacturers to display their process certification marks. The third method consists of auditing the manufacturing management standards, such as ISO 13485 for medical devices, and assuring the manufacturer meets that standard. The final way uses accreditation by an authoritative body in order to recognize that a certain organization or person is capable of carrying out a certain task. For example, in Europe, Notified Bodies are accredited to carry out conformity assessments of all medical devices. [4]
Good Manufacturing Practice (GMP)
Manufacturers usually establish predefined quality systems and follow them to help consistently ensure products and to meet the applicable requirements and specifications. These requirements and specifications are known as Good Manufacturing Practice (GMP), which has lately been referred to as “current GMP” due to modifications made to the old version of GMP. Current or not, GMPs are regulations that describe the methods, equipment, facilities, and controls required for producing medical devices. [6]
1976 was the first year GMPs as standards were applied to medical device manufacturing through a set of procedures and policies, referred to as a quality system. GMP was established for medical devices due to the existence in the market of inconsistently made medical devices that were causing injury when used. [7]
GMPs were initially enforced in the United States by the FDA whereas, in the European Union, GMP inspections are performed by National Regulatory Agencies. In Australia, GMP inspections are performed by the Therapeutic Goods Administration (TGA) while, in India, they are performed by the Ministry of Health. Worldwide, GMP inspections are performed by similar national organizations. Each of the inspectorates carries out routine GMP inspections to ensure that products are produced safely and correctly. Additionally, many countries perform Pre-Approval Inspections (PAI) for GMP compliance prior to the approval for marketing of a new product.
The main goal of the GMP system is to achieve a loop for control and feedback so that manufactured goods sold to consumers are safe and perform their intended functions. There can be no doubt that this goal satisfies everyone involved in the production of medical devices [7]. This loop can be achieved by assuring that manufacturers are following the manufacturing process and that the finished products are safe and functioning properly as well as by assuring that manufacturers are using the correct instructions and drawings or that validated designs are working for their intended uses with stress condition.
Moreover, GMP regulations deal with issues such as personnel qualifications, recordkeeping, sanitation, cleanliness, equipment process validation, verification and complaint handling. Most GMP requirements are wide ranging and open ended, and allow manufacturers to make decisions individually about the best implements to use and the necessary controls. This feature provides a great deal of flexibility and requires manufacturers to interpret the requirements in a way that makes sense for all parties involved. [1]
Medical Devices Classification
Even though the classification of medical devices is not uniform worldwide, the same concept of classification is followed. Medical devices classification is considered to be a risk-based classification. This means that, based on the risk and potential hazards the devices pose to public health and individual patients, manufacturers and regulatory authorities can classify medical devices between class I to class III where a higher class signifies a more critical and risky device. [8]
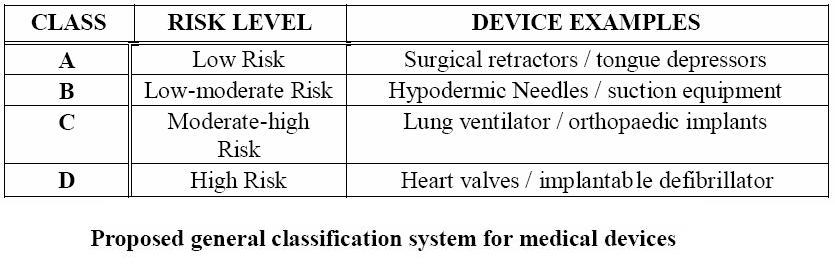
Medical devices classification exists for a variety of reasons. Firstly, they allow recognition of the differences in the degrees of inherent risk. Additionally, they establish conformity assessment requirements based on risk level of the devices. They also establish conformity assessment process options also based on device’s risk level. Finally, they allow for regulatory controls to be proportional to risk. [2]
The Global Harmonization Task Force (GHTF) created 16 rules for medical devices classification. These rules cover major areas including non-invasive devices, invasive devices, active devices and additional rules specific to public health considerations. These 16 rules are illustrated in what are known as decision trees.
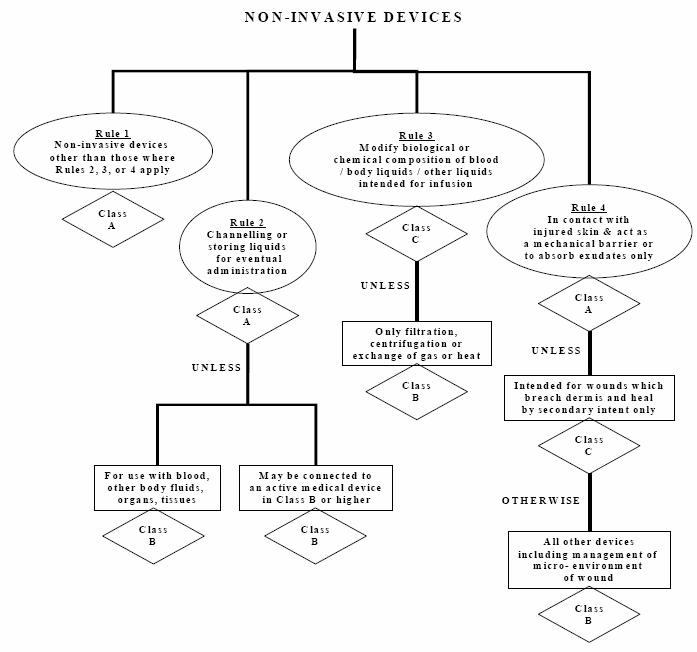
As an example, the FDA classifies medical devices into three classes based on their associated risk. Class I medical devices have the lowest regulatory control because they pose the minimum possible harm to the user. Class I devices are normally simple in design and manufacture and have a safe history of use. Most class I devices are exempt from premarket notification and may be exempt from compliance with the good manufacturing practices regulation. Class II medical devices are devices where the general controls are not sufficient to assure their safety and effectiveness, so existing guidance documents are available to provide assurances of safety and effectiveness. In addition to compliance with general controls, class II devices are required to comply with special controls, which include special labelling requirements, mandatory performance standards both internationally and in the United States, post-market surveillance and, finally, FDA medical-device specific guidance [8]. Furthermore, class II devices typically require pre-market notification by submission and FDA review of a 510(k) clearance for market submission. Class III medical devices are considered to have the strictest regulatory controls. For class III medical devices, sufficient information is not available to assure safety and effectiveness through the application of general controls (as for class I) and special controls (as for class II). Class III devices usually support or sustain human life, are of extensive importance in preventing harm to human health, or present a potential unreasonable risk of illness or injury to the patient. To market a class III medical device, a pre-market approval (PMA) submission to the FDA is required. [9]
Medical Device Safety and Risk Management
There are several essential elements in ensuring the safety of medical device and deriving the optimum level of benefits from such devices. The elements to be considered in this respect are:
- it is not possible to guarantee an absolute safety level with medical devices as there is always a certain degree of risk associated with any medical device,
- medical device safety is a risk management issue,
- the effectiveness and the performance of the medical device largely determine the degree of risk associated with the device,
- the risk management should cover the aspects spreading throughout the life span of the device and
- the responsibility for safety needs to be shared among all the stakeholders.
Obviously, all medical devices carry some degree of risk, and this risk can cause complications in specific circumstances. It is often the case that, until the device gains experience on the market, the specific types of problems that will arise are undetectable. For example, an implantable medical device might fail—without its failure being predicted at the time of implantation; however, this failure may only be reproduced in situations that are exclusive to certain patients. In fact, the present approach to medical devices safety is to maximize the estimation of the probability of the device becoming a hazard. This estimation is called risk assessment[4]. Medical devices manufacturers should create and follow a risk management process while developing a new product or when applying changes to an existing one [10]. However, the results of the risk management process need to be reviewed by the FDA in order to determine whether the device is safe or not.
Risk management is considered to be a necessary skill, and all biomedical engineers are required to have a complete understanding of how to use it consistently. Risk management involves combinations of procedures, practices and management policies that deal with the responsibilities of identifying, analysing, controlling and monitoring risk [11]. Given that hazard is defined an adverse event’s potential source of danger, risk is a measure for the combination of hazard, adverse event occurrence, and its severity or overall impact. Risk assessment comprises two major sections: the risk analysis and the following risk evaluation. Risk analysis involves the identification of all possible hazards, and risk evaluation is the estimation of the risk associated with each hazard. Generally, personnel in the risk assessment profession need to have various skills, such as experience and verification, computational and even estimation abilities. Many factors can affect the assessment of risk, including personal perception, culture, economic situations and political atmospheres. [4]
The term ‘risk’ thus signifies the probable rate of occurrence of a hazard which results in harm or increasing the degree of severity of the harm. Therefore risk as a concept has the elements of
- the possibility of an event creating a hazard
- the severity of the consequence of such event resulting in the harm.
There are different probability levels at which risks can occur. The US Food and Drug Administration have prescribed three levels of risk – major, moderate and minor which approximately indicate critical, marginal and negligible levels respectively. Poor manufacturing or insufficient attention to the design of the elements affecting the performance of device can present potential hazard. A typical risk management process would include
- developing strategies to deal with risks identified as associated with the device,
- monitoring the risks associated,
- invoking a contingency plan to mitigate the risks,
- managing the crisis resulting from the risks, and
- identifying the ways to recover from the crisis
Risk management usually begins with the development of the design input requirements. As the design is developed, the identification of new risks should take place. To systematically identify and eliminate these risks, the integration of risk management with the design process should occur in the early stages. In this way, all undesirable risks will be identified and managed early on, and as a result, changes will be easier to make with fewer expenses. [11]
In classifying medical devices there is the need that the potential areas of hazard that warrant consideration are the degree of invasiveness of the device and the duration of the contact of the device with the patient or the user. The risk classification also considers the body system affected and the local versus systemic effects of the device under question. It is to be noted that an invasive device is likely to have higher potential hazard as compared to a device that is non-invasive. Similarly the devices which are in contact for longer duration with the vital organs of the body like heart or the great arteries may cause higher potential hazard to health and safety of the patients. The devices which tend to have systemic effects on the users are also considered as high risk potential devices. Consequently these devices are to be classified as carriers of higher classes of potential hazard or risk. The degree of regulatory control imposed on any device therefore is framed based on the proportion of the potential risk the device carries. This approach is generally described as ‘risk management’. This is evident from the recommendation of Global Harmonization Task Force (GHTF). This recommendation is contained in the first requirement of the “Essential Principles of Safety and Performance of Medical Devices” which states that:
“Medical devices should be designed and manufactured in such a way that, when used under the conditions and for the purposes intended and where applicable, by virtue of the technical knowledge, experience, or training of intended users, they will not compromise the clinical condition or the safety of the patients or the safety and health of users or where applicable other persons provided that any risks which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety”
From this statement it may be observed that an assessment of the potential risks against the likely benefits from the medical devices is of paramount important to classify the device to a particular risk class. The objective here is to maximize the benefits and minimize the associated risks. The manufacturers of medical devics also follow the same risk management approach while designing their equipments and devices. It is with this view in mind that the International Organization for Standardization (ISO) has framed the global standard of ISO 14971-2000 which provides the manufacturers a framework which includes risk analysis, evaluation and control for risk management in designing, development and manufacturing the medical devices. This standard also prescribes for the monitoring of the safety and performance of the device even after sale during the use of the device by the patients or other users [4].
The main purpose of classifying the risks associated with medical devices is to ensure that the regulatory controls that are applied to medical devices are framed and implemented in proportion to the potential risk being carried by the devices. Various methods of assuring compliance with regulatory controls have been prescribed by the Statutory Conformity Assessment Authority. However there is no harmonization has been achieved in prescribing the regulatory rules in respect of different classes of devices because of the fact the Regulatory authorities are under an obligation to reflect the local needs or other social considerations at the time of introducing new regulations or classifications in respect of new devices being introduced in the market.
In general with the increase in the level of risks associated with the device there would be corresponding increase in the level of regulatory requirements. The regulatory controls would normally take into account
- the operation of a quality system
- technical and operational data of the device concerned
- results of the tests conducted on the product using either in-house facilities or other independent outside resources,
- the documentation of clinical evidence produced by the manufacturer in support of his claims,
- the necessity for and the frequency of any independent external audit to be carried out on checking the quality system of the manufacturer and
- an independent external review of the technical details submitted by the manufacturer in respct of the new device proposed to be introduced by him.
In general risk management activities are purported to identify the opportunities to improve the performance of medical devices. Since there is always the need for human interaction in putting the medical device into use, the element of risk necessarily has to be evaluated and minimizing the level of routine human interaction would result in improved efficiency and reduction in the degree of risks associated with the device. The following figure illustrates a general concept regarding the increase in regulatory control with increase in the risk associated with risk.
Conceptual Illustration of Regulatory Controls
Increasing with Device Risk Increse Class
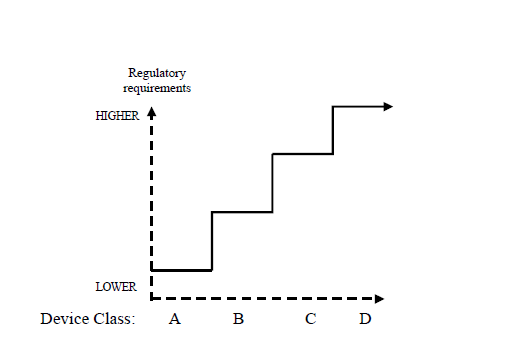
Before beginning to implement risk management, it is important to determine the hazard severity levels and their probabilities of occurrence. Then it is important to determine the risk acceptance criteria by categorizing each risk. The following tables provide an example of these steps:
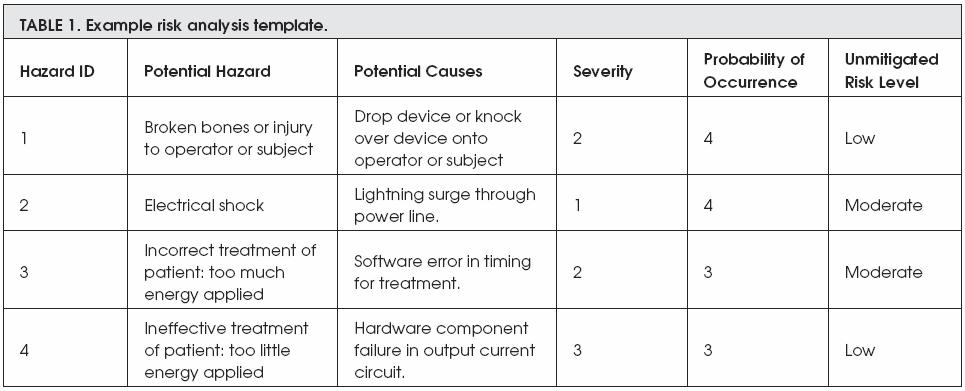
Examble 1 shows four different potential risks that might be occured during the use of a medical device. This table detailed each risk by including the following:
- Hazard ID which is an ID that given by a manufacturer
- Potential Hazard which is the description of the risk that may occur
- Potential Causes means the reason behind the risk occurance
- Severity: from scale 1 to 3, how sever is that risk (as in examble 2)
- Probability of Occurance: from scale 1 to 5, how often is the risk occur (as in examble 3)
- Unmitigated Risk Level: the general or overall description of the risk
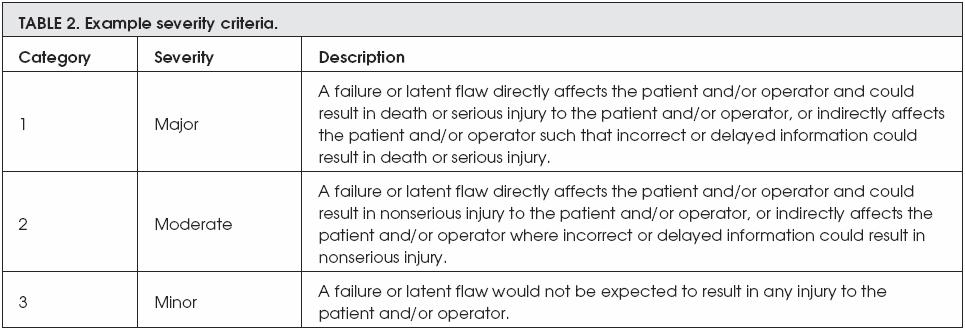
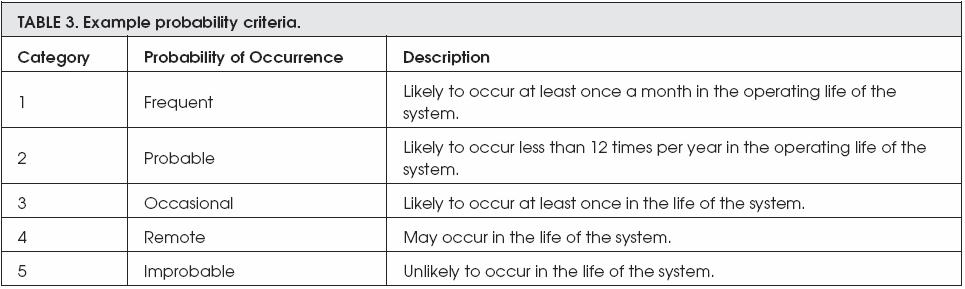
However, the next two exambles are describing both the severity criteria and the probability of risk occurrence. The severity of any risk is devided into three different categories; these categories are major, moderate, and minor, whereas the probability of risk to be occurred is devided into five categories; these categories are frequent, probable, occasional, remote, and improbable.
On the other hand, ISO 14971:2007 is a standard dedicated to the quality management and corresponding general aspects of medical devices, and covers risk management for in vitro diagnostic (IVD) medical devices. Moreover, it specifies a process for the manufacturer to follow in order to identify the hazards associated with medical devices, including IVD medical devices. In addition, it covers the estimation and evaluation of risks associated with the manufacturing process. Furthermore, it controls these risks and monitors the effectiveness of the controls. [12]
Clinical Trials for Medical Devices
Ethical guidelines require that many steps be performed in order to prepare medical devices before they enter the market. After experiments of the device have been conducted in animal studies, the device must be further tested in a clinical study on human subjects. Furthermore, different key steps enter into the design and execution of human-subject studies so that they may be used to support submissions to the regulatory authorities for the pre-market approval.
The main goal behind the performance of clinical trials is to provide an evaluation of the safety and effectiveness of the medical device and to ensure it fulfils the manufacturer’s claims. If anything is found in the design that can affect the medical device’s safety and effectiveness, all regulatory authorities have the legal right to immediately reject that product. However, manufacturers should be aware that both time and cost are associated with designing, conducting and analysing medical devices’ clinical trials, so they must take care to balance production against the delay in approving studies. [13]
Particularly when designing clinical trials, manufacturers should be attentive to and control the known or suspected sources of bias and errors. A bias refers to characteristics of the investigator or study population that interfere with the accuracy of measuring the variables, whereas an error signifies the failure to accurately measure the variables. [13] The first step to designing a good and efficient clinical investigation is to predefine a clear objective and form it into a research question. In answering those questions, researchers should be able to identify and select the variables and expend as much effort as possible to keep the investigation balanced and controlled. Afterwards, they should choose an appropriate study population targeted for the medical device’s application. [13]
Additionally, good clinical trials should have a suitable and detailed protocol that should be submitted to the regulatory agency along with submission file. After the protocol has been defined, the clinical study can be conducted and the results analysed. Based on those results, the regulatory agency will be able to decide whether to approve or reject the product depending on in which country the submission is taking place. [14]
Background on TMS, DBS and ECT
Many devices on the market are considered to be high-risk products. Due to the urgent need for technology, however, some of those devices already on the market are still in the process of obtaining the appropriate approval from regulatory agencies, while some have already been approved. In this chapter, there will be a discussion about some critical medical devices that apply energy to the human body. This discussion will include an introduction and background of the devices, details of their regulation, and what devices have been previously approved and where.
Transcranial Magnetic Stimulation (TMS)
TMS is a new technique used as in non-invasive therapeutic and diagnostic applications in neurophysiology research and clinical applications. TMS is assumed to have advantages over electrical stimulation applied to the deep nerves and brain without discomfort and corporal invasion. In terms of its applications, single-pulse TMS is widely used to study the motor and visual systems. Another type of TMS is repetitive TMS (rTMS), which produces greater effects on the brain and the brain’s functions. [15]
History and background
The idea to use TMS began with the use of eddy currents in inductive brain stimulation in the 19th century. However, the first TMS study only took place in 1985 by Anthony Barker in Sheffield, England. At that time, its application was involved in the demonstration of the conduction of nerve impulses from the motor cortex to the spinal cord. A few years earlier, this had been performed using transcranial electrical stimulation, but scientists found it caused severe discomfort. By stimulating different points on the cerebral cortex and recording such responses as muscle movements, maps of functional brain areas may be obtained. When using MRI or EEG, information may be obtained about the cortex and about area-to-area connections.
Principle of operation
TMS and repetitive TMS (rTMS) have been used for almost 20 years for the noninvasive study of the human brain. TMS is generally used as a diagnostic tool to detect the relations between excitatory and inhibitory circuits inside precise cortical areas of the brain. In recent studies, it has been considered an effective therapeutic tool for diseases such as depression, Parkinson’s and other neurological conditions. [17]
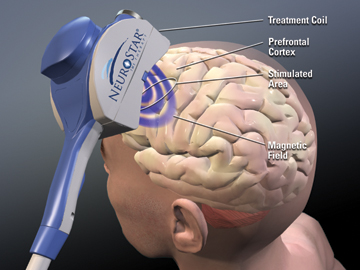
TMS non-invasively excites neurons in brain. A weak electric current is induced in the tissue by rapidly changing magnetic fields, known as electromagnetic induction. In this way, we can trigger brain activity with minimum discomfort and study the brain’s functionality and connectivity. [16]
Before describing how TMS works, it is important to differentiate between TMS and rTMS. In terms of operation, a TMS pulse causes a population of neurons in the neocortex to depolarise and discharge an action potential. When applied to the primary motor cortex, this produces a motor-evoked potential, which can be recorded using EMG. If applied to the occipital cortex, the subject detects flashes of light. When applied to other parts of the cortex, patients do not consciously experience any effect, but their behaviour might be slightly changed and the brain activity could be detected using PET-CT or fMRI. However, these effects do not outlast the period of stimulation. [17]
On the other hand, rTMS produces effects that last longer than the period of stimulation. In addition, rTMS can increase or decrease the excitability of corticospinal or corticocortical pathways depending on the intensity of stimulation, coil orientation and frequency of stimulation. The mechanism of these effects is not clear, although it is widely believed to reflect changes in synaptic efficacy akin to long-term potentiation and long-term depression. [19]
Clinical applications
In terms of applications, TMS is used clinically in order to measure the activities and functions of specific brain circuits. Its widely accepted use is to measure the connection between muscles and the primary motor cortex. This is considered to be useful in the treatment of stroke, multiple sclerosis, spinal cord injury and motor neuron disease. In addition, brain plasticity can be measured using rTMS, and it is believed that this abnormality is the cause of many brain diseases. In addition, TMS is used to treat other diseases such as tinnitus, Parkinson’s disease, dystonia, amyotrophic lateral sclerosis, epilepsy, migraine, dysphasia, hemispatial neglect, clinical depression, phantom limb and chronic pain. [16], [20], [21].
TMS and Depression
Patients who are suffering from depression disorders sometimes do not respond to some of the regular antidepressant medications. Some recent studies have appeared and are assumed to have confirmed that standard left-sided rTMS has antidepressant efficacy, but that the degree of clinical effect may be somewhat limited. Promising data are emerging suggesting that other approaches, including right unilateral rTMS and sequential bilateral stimulation, may have equal or potentially greater effects. [22]
According to [23], 24 patients with major depression and stable medication received high frequency (10 Hz) rTMS over the left dorsolateral prefrontal cortex (DLPFC) for two weeks. The results indicated that, after two weeks of rTMS, a mean reduction of 30% of the initial Hamilton Depression Rating Score (HAMD) was observed. This means the former response to antidepressant agents proved significant and that the high pre-treatment anterior cingulate activity and low treatment resistance to pharmacologic therapy were positive predictors for a treatment response to rTMS.
TMS and Epilepsy
Brain stimulation has been receiving increasing amounts of attention as an alternative therapy for cases of epilepsy that cannot be treated by either antiepileptic medication or surgical resection of the epileptogenic focus. Although TMS is the simplest and least invasive procedure that can be performed on a patient, it has some limitations regarding its effectiveness on epileptogenic areas (mesial temporal structures), which lie too deep beneath the skull surface. However, a few rTMS experiments have been performed in animal models of epilepsy. One of those studies has proven that low frequency stimulation at 0.5 Hz increased latency to onset of pentylenetetrazol-induced seizures in rats. [24]
In addition, there have been scattered reports of the use of rTMS to treat patients with seizures, or cortical myoclonus. In an open study, eight of nine patients with complex partial seizures and temporal lobe epilepsy were reported to have a mean seizure reduction of 38.6%, comparing four weeks before and four weeks after five days of 0.33 Hz rTMS [24]. When the cost effectiveness of this percentage will have been calculated, rTMS will become a useful tool to treat epilepsy.
TMS and Parkinson’s disease
Functional neuroimaging provides insights into the pathogenesis of motor symptoms in Parkinson’s disease and improves the understanding of both established neuromodulatory therapies such as deep brain stimulation (DBS) and potential ones such as rTMS, which has been suggested as becoming a potential non-invasive alternative for neuromodulation of cortical function. Although rTMS is not currently a treatment, recent rTMS studies in Parkinson’s disease suggest its promise and illustrate how functional imaging can guide rTMS application. Also, they suggest that subcortical dopamine release could be an rTMS mechanism of action, which, of course, will enhance the way in which patients with Parkinson’s disease are treated. [21]
Risks of transcranial magnetic stimulation
Even though TMS is a non-invasive application, it does come with the risk of undesirable side effects, such as an increased possibility of seizure. Because of this risk, some international organizations advise that the procedure should be performed only when medical assistance is available, in case of emergency, and that users who administer it must be able to provide such assistance. In addition, there are some risks associated with TMS such as light-headedness, headache, tingling and facial muscle contractions. [25]
A study of 301 medication-free patients with major depression who had not benefited from prior treatment was performed to study the safety and efficacy of TMS. This study showed that active TMS was significantly superior to sham TMS on the Montgomery-Asberg Depression Rating Scale (MADRS) at week 4 (with a post-hoc correction for inequality in symptom severity between groups at baseline), as well as on the Hamilton Depression Rating scale (HAMD)—HAMD17 and HAMD24 scales at weeks 4 and 6. Response rates were significantly higher with active TMS on all three scales at weeks 4 and 6. Remission rates were approximately doubly higher with active TMS at week 6 and significant on the MADRS and HAMD24 scales (but not the HAMD17 scale). Active TMS was well tolerated with a low dropout rate for adverse events (4.5%) that were generally mild and limited to transient scalp discomfort or pain. This means TMS is effective in treating major depression with minimal side effects reported and may be assumed to offer clinicians a new alternative in the treatment of depression. [26]
H-coil is a new development in TMS research that allows direct stimulation of deeper neuronal pathways than does the standard TMS coil. A study assessed the possible health risks and some cognitive and emotional effects of two H-coil versions designed to stimulate deep portions of the prefrontal cortex, using several stimulation frequencies. This study proved that there are no adverse physical or neurological outcomes, and the computerized cognitive tests found no deterioration in cognitive functions, except for a transient short-term effect of the H1-coil on spatial recognition memory on the first day of rTMS. Accordingly, H-coils offer a new, safe tool with potential in both research and clinical applications for psychiatric and neurological disorders associated with dysfunctions of deep brain regions.
TMS and regulatory agencies
In terms of regulatory clearance, the only cleared TMS system is the Neuronetics, Inc. TMS system, NeuroStar TMS, which was just had the clearance by the FDA in October 2008. Notice that this is only clearance and not considered as a final approval[27]. However, there are a few manufacturers who manufacture TMS, and all those manufacturers are in the submission process to the major regulatory agencies.
Case study 1: Neuronetics TMS therapy system
Neuronetics is a U.S. company that specializes in non-invasive therapy to treat neurological disorders. Their TMS machine is called NeuroStar. Neuronetics claims that their 40-minute outpatient procedure, which does not require sedation or anaesthesia, is clinically tested and that their studies show that the efficacy and safety in treating depressed patients who have had an inadequate response to previous therapies has succeeded. [18]
The FDA, after reviewing the Neuronetics file with their submission for approval, finally, cleared the NeuroStar TMS system on October 8, 2008. The safety and effectiveness of the NeuroStar system was evaluated in a clinical trial consisting of three phases identified as study 01, study 02 and study 03. Study 01 was designed as a multi-centre, randomized, parallel-group, sham-controlled clinical trial to demonstrate the safety and effectiveness of the device for subjects diagnosed with DSM-IV-defined major depression, which did not benefit from prior adequate treatment with oral antidepressants during their current major depressive episode. Study 02 was an open-label clinical phase for subjects who had received either active treatment or sham treatment in Study 01. Subjects in study 02 were considered non-responders based on pre-defined criteria for response to treatment in study 01. Study 03 was an open-label, uncontrolled clinical trial designed to demonstrate the durability of rTMS treatments. Subjects from Study 01 and/or Study 02, who were considered responders based on pre-defined success criteria, were followed for six months while receiving oral antidepressant monotherapy. [18]
Case study 2: Brainsway TMS system
Brainsway is an Israeli manufacturer dedicated to manufacturing TMS systems. Brainsway has performed clinical trials on healthy volunteers and claim they have proven the safety of their technology and its ability to stimulate deep brain regions. They are in the process of clinically testing their system on patients suffering from major clinical depression and with patients suffering from schizophrenia. All their clinical trials are being conducted in full accordance with FDA guidelines. [28]
Brainsway has performed two different safety and efficiency studies and three different clinical trials. The clinical trials concerned depression, schizophrenia and bipolar disorders, and the results for all three trials were considered positive. [28] Brainsway believes these results will allow them to arrange for an application for the CE mark to enter the EU market and then continue with a similar application to the FDA in order to enter the U.S. market. [29]
Some clinical trials and safety tests on TMS
According to Neuronetics’ study [30], the response of TMS in patients with major depression was successfully maintained during the transition to continuation therapy. In addition, for active TMS patients continuing into the maintenance-of-effect study, Kaplan-Meier relapse rates at 24 weekly follow-ups ranged between 5.6% and 22.2% depending on the relapse definition used.
This study program has consisted of three separate clinical protocols with 301 patients: a six-week randomized controlled trial of active TMS (Neuronetics model 2100 system versus sham TMS), a six week open-label extension study for non-responders in the first trial, and a 24-week continuation pharmacotherapy maintenance-of-effect study for responders in either the controlled or the open-label study. The treatment parameters were optimized in a fixed, maximum-achievable dose design.
In sum, the acute response to the TMS shows a persistence of positive benefits in both short-term transitions to continuation therapy and after long-term follow-up. This proves the effectives of TMS in the treatment of patients with major depression who did not receive any adequate benefit from previous treatment.
On the other hand, there are many doubts about the maximum energy that can be delivered to the patients without negatively affecting the brain. There is still a shortage of experiments on human subjects that study the effects of higher energy delivered by TMS. When dealing with animal subjects, according to [31], it is important to control the intensity of the magnetic energy delivered by TMS in order to maximize the safety and efficacy of the treatment. In that study, a group of researchers were investigating the effects of TMS on the brain by focusing on long-term potentiation (LTP) in the rat hippocampus. Their conclusions suggest that the synaptic plasticity in the hippocampus was impaired by strong TMS.
Deep Brain Stimulation (DBS)
DBS can be defined as a surgical treatment that involves the implantation of a medical device called a brain pacemaker. The brain pacemaker sends electrical impulses to specific parts of the brain. DBS with the ability to select brain regions provides therapeutic advantages for otherwise treatment-resistant movement along with affective diseases such as Parkinson’s disease, chronic pain, tremor and dystonia. Even though DBS has a long history, its unknown principles and mechanisms are still unclear. In a controlled manner, DBS can directly apply changes in brain activity.
History and background
DBS is an interesting example of the relationship between clinical science and basic science as a two-way process. The first report of DBS being used in the subthalamic nucleus to treat Parkinson’s disease was published in 1993. Benabid’s clinical group in France had first used DBS in the thalamus in 1987 with three patients. This experiment involved implanting electrodes in the two sides of brain. Until now, this has been the standard approach for the use of DBS in Parkinson’s disease patients. [33, 34]
The FDA approved DBS as essential tremor treatment in 1997. In 1998, the FDA approved it for Parkinson’s disease and in 2003 for dystonia[35]. DBS is also used as treatment for chronic pain, and it has been used as treatment for various affective disorders such as clinical depression. [32]
Principle of operation
The DBS system consists of three parts: the implanted pulse generator, the lead and the extension. The battery-operated implanted pulse generator placed in the brain produces pulses of energy that block the abnormal activity located in the brain that result in movement disorders like dystonia. [32]
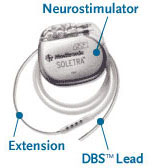
DBS leads are fixed in the brain based on the type of symptoms that need to be addressed. In cases of non-Parkinsonian essential tremor, it is important to place the lead in the ventrointermedial nucleus in the thalamus. However, for dystonia and symptoms related to Parkinson’s disease such as rigidity, akinesia and tremor, the lead can be placed in either the subthalamic nucleus or the globus pallidus. [32], [37]
To implant a DBS device, a surgeon drills two holes in the top of skull, implants the two electrodes deeply in the brain, runs wires under the neck skin and finally connects them with a device that looks like pacemaker that needs to be implanted under the skin in the chest. [38]
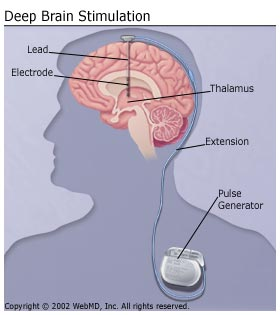
All three components are surgically implanted inside the body. The left side of the brain is stimulated to address symptoms on the right side of the body, and the right side of the brain is stimulated to address symptoms on the left side of the body. However, the success of DBS surgery is directly related to being able to locate the specific area in the brain for stimulation. [35]
Clinical applications
There are a variety of applications that can be performed by DBS; however, the main two conditions that are treated with DBS are tremor and Parkinson’s disease. In addition, it is assumed that DBS has beneficial effects in treating depression, Tourette syndrome, obsessive-compulsive disorder and dystonia, but this benefit is confounded by the fact that it takes weeks to months to be manifested. Another problem lies in the fact that it is not clear which electrode geometries, pulse durations, stimulation amplitudes and frequencies are most efficient for these new therapeutic applications. [40], [32]
DBS and Parkinson’s disease
Parkinson’s disease is a neurodegenerative disease and known to cause symptoms such as rigidity, tremor, bradykinesia and postural instability. It is important to recognize that DBS does not cure Parkinson’s, but it does help to control some of its symptoms and thereby enhance the quality of the patient’s life. Nowadays, the procedure is only given to patients whose symptoms cannot be controlled with medication or to patients experiencing complications and side effects from medication. The direct effect of DBS on the brain cells’ physiology and on neurotransmitters is currently being debated, but, by sending electrical impulses in high frequency form into specific parts of brain, DBS can reduce the symptoms and side effects introduced by medications for Parkinson’s, which thereby leads to a decreased reliance on medication. [32]
DBS and Depression
Some scientists believe that the use of DBS for depression should be a last resort and only for the most desperate patients. This is due to the complication of the implantation procedure. These scientists believe that, in the majority of cases, it is preferable to use electroconvulsive therapy (ECT) in stages before resorting to DBS. [38]
The first trial of DBS for depression was performed in Toronto, Canada, in 2005. In this trial, researchers implanted Medtronic DBS for six patients. The results showed that five patients responded well directly after the treatment, and four of those five are still doing well, up to two years after. However, the team leader is still uncertain if the positive result was due to the device or because the patients had independently recovered from the depression symptoms. [38]
DBS and Dystonia
Dystonia can be defined as an involuntary movement disorder that manifests as muscle contractions that lead to abnormal postures and spasms, which affects body-emotion performance. The failure of traditional medication in many patients with severe forms of dystonia has led to neurosurgeons’ renewed interest in changing their approach to treatment. Recently, DBS has shown how much easier and effective the treatment of dystonia can be, particularly for patients who are suffering from primary generalized dystonia. [41]
Risks of deep brain stimulation
Even though the extent of DBS’ helpfulness has been shown for some patients, the potential for neurological side effects still exists. There are many reports in the literature that show the possibility of complications such as hypersexuality, hallucinations, apathy, cognitive dysfunction, compulsive gambling and depression. Despite the temporary nature of these side effects, they must still be considered because they may lead to further complications. [32]
Furthermore, there is the possibility that, during surgery, the electrodes become displaced or dislodged due to slight shifting of the brain. This may cause complications similar to personality changes. While electrode misplacement is considered easily identifiable using CT or MRI, it must be remembered that other complications may arise during surgery, such as bleeding inside the brain. [32]
Surgical complications can be very serious and may depend on the degree of the surgeon’s experience. The most important adverse event that may occur is brain hemorrhage resulting in symptoms similar to those of stroke and possibly leading to death; however, this complication rarely occurs (less than 1%). Additionally, insufficient cognition and behaviour may occur in elderly patients known to have cognitive impairments. A careful preoperative screening will help minimize this problem. Both severe depression and suicide have been reported after DBS implantation, occurring in 4.3% of patients, and linked to a history of depression. [37]
DBS and regulatory agencies
There is only one supplier who manufactures DBS devices: Medtronic. Medtronic has a long history of experience with regulatory requirements, especially with FDA in the United states. Currently, Medtronic is trying to find a way to get FDA approval for their DBS Activa, and the FDA has approved it to treat Parkinson’s and to treat dystonia but under the Humanitarian Device Exemption. The FDA is considering DBS a class III medical device because of its intended use as an implantable medical device. [42], [43]
Case study 1: Medtronic DBS therapy
Medtronic has been developing brain stimulation technology since the late 1980s. In 1987, professors from Grenoble University in France published the outcome of the first use of brain stimulation for the treatment of movement disorders. Since then, three indications for DBS therapy (called “Activa” by Medtronic) have become commercially available. These are Activa Tremor Control Therapy, Activa Parkinson’s Control Therapy and Activa Dystonia Therapy. Activa Tremor Control Therapy, which was approved in Australia, Canada and Europe in 1995 and approved by the FDA in 1997, is brain stimulation that targets the thalamus with electrical stimulation in order to suppress tremor associated with either Parkinson’s disease or essential tremor. Alternatively, Activa Parkinson’s Control Therapy, which was approved in Australia, Canada and Europe in 1998 and by the FDA in 2002, is brain stimulation that targets the internal globus pallidus or the subthalamic nucleus with electrical stimulation in order to suppress some of the motor symptoms of advanced, levodopa-responsive Parkinson’s disease. Finally, Activa Dystonia Therapy, which was approved by the FDA in 2003 under a Humanitarian Device Exemption, is brain stimulation that targets the internal globus pallidus or the subthalamic nucleus on one or both sides of the brain with electrical stimulation to relieve the muscle contractions that characterize certain types of dystonia. [44]
Some clinical trials and safety tests on DBS
Medtronic, as the only manufacturer in the market of a DBS system, uses electromagnetic compatibility (EMC) protective measures that are similar to those in pacemakers. In the absence of established EMC standards for neurostimulation, Medtronic has based EMC immunity testing on the established standards for EMC testing of pacemakers. The pacemaker testing requirements define the EMC test frequencies and field strength levels to which patients implanted with cardiac pacemakers may be exposed. It is expected that a patient implanted with a neurostimulation system can be exposed to the same type of EMC environment. [45]
According to [45], Medtronic has performed two different clinical trials on their Activa product. The first trial concerned the safety and effectiveness of using DBS for the treatment of Parkinson’s disease, and the second trial concerned the use of DBS for the treatment of tremor. The Parkinson’s disease clinical trial was compromised of two parts: safety and effectiveness. In the DBS effectiveness study, 102 patients out of 117 showed improvement in Parkinsonian symptoms. Additionally, three patients died during the 12 months of the clinical trials, with the causes of death being esophageal neoplasia, myocardial infarction and late stages of Parkinson’s disease. However, these causes of death were completely unrelated to the implantation of the DBS system.
Electroconvulsive Therapy (ECT)
Electroconvulsive therapy (ECT) has several alternative names including shock treatment, shock therapy, electroconvulsive shock treatment, electroshock treatment and electroplexy. According to [46], the number of electroconvulsive therapy treatments given per day in the U.S. was estimated as 10,000 in the early 1970’s. At that time, depression was the most common illness treated with ECT. ECT is used to treat suicidal manic depressives and acute schizophrenics.
The essence of the treatment is the convulsion or seizure rather than the electrical stimulation of the brain. In fact, convulsions induced by the injection of drugs or the inhalation of chemicals have approximately the same therapeutic effects as ECT. [46]
History and background
Approximately 70 years have passed since Von Meduna first attempted to produce therapeutic convulsions in patients with mental illness via the injection of camphor oil. Many successive advances in technique have led ECT to become comparable with other medical procedures in terms of safe anaesthetic acceptance, especially for those who undergo general anaesthesia, and, yet, the technique remains controversial. The reasons for this are unclear, but a few suggest themselves—these reasons become evident when comparing ECT with other procedures that have been used in psychiatry, and some are related to side effects produced by electrical shock. The most obvious reason is the lack of information about the exact mechanism of the procedure. [47]
Until recently, ECT has suffered from a bad reputation, particularly in the press and the public eye. The reason for this can be traced to the fact that, until the beginning of the 1950s, ECT was applied without anaesthesia or muscle relaxation drugs, which resulted in violence and difficulty in controlling patients due to the shock of the seizures that might have caused the patient to harm or injured themselves. [47]
Principle of operation
When a decision is made for a patient to have ECT, a pre-evaluation judgment should be taken in consideration, with the purpose of determining the amount of energy and the energy level that needs to be delivered to the patient. After this and prior to the treatment, patients should receive a measured amount of anaesthesia and muscle relaxation drugs. In some countries, however, physicians are still using ECT procedures without anaesthesia or muscle relaxation drugs that have been banned by the WHO. [47]
Next, the shock delivery process involves the placement of the electrode or electrodes on the patient’s head, depending on the type of procedure to be performed. The electrode(s) actually deliver an electrical stimulus at levels recommended to be higher than the individual’s seizure threshold because, below these levels, treatment will not be effective. [48]
Clinical applications
According to [47], ECT is the most effective treatment for major depression. Furthermore, when compared to antidepressant drugs, studies have shown that ECT is more effective. Also, some studies have shown that the use of ECT decreases the suicide rate in people with depression. ECT is an also effective anti-manic treatment and could be lifesaving for patients with acute manic excitement who are at high risk of cardiovascular collapse and death. [47] ECT is also effective in treating functional psychosis developing after the age of 60 to 65. In addition, ECT is effective in the treatment of coarse brain disorders and systematic illnesses that affect brain function. [47]
In general, after 40 years of practice, ECT seems to be effective when properly administered in the treatment of a wide variety of mental disorders [47]. Following are some details about some of ECT’s clinical applications.
ECT and Depression
Since the early 1950s, many studies have been performed with the majority of researchers supporting the efficacy of ECT in the treatment of severe depression. Many patients who suffer from depression do not respond to medications given as antidepressants, or, because of the dangerous side effects caused by the antidepressant, some patients avoid that kind of treatment. Therefore, they find themselves moving toward the use of ECT, which has been proven to be effective [49].
One study has shown that ECT has at least the same effect of antidepressant medications when dealing with major depression [49]. Another has shown that the efficacy of ECT is superior to medications in severe depressive illness [46]. Furthermore, according to [50], rTMS is not as effective as ECT in the treatment of depression. In fact, of the roughly 100 different books and articles examined for this thesis, almost all of them support the efficacy of ECT in the treatment of depression.
ECT and Mania
The rapid spread in the use of lithium therapy (as an old method for treating mania), at a time when the importance of controlled trials of ECT was also being recognized, had an inhibiting effect that allowed scientists to begin researching the efficacy of ECT on mania. [47]
Several retrospective studies have shed light on ECT and Mania. For instance, a review of all patients receiving ECT at McLean Hospital between 1973 and 1986 showed that 57% of the manic patients improved significantly or recovered after receiving ECT [47]. In addition, according to a chart-review study that compared manic patients who received ECT with patients who were hospitalized for drug treatment of mania, 96% of the ECT patients had been discharged from the hospital compared with only 44% of the other group [47].
Though a few studies suggest ECT’s efficacy on mania, a great deal of effort still needs to be applied to further research on manic patients to ensure ECT is a safe treatment for them.
ECT and Schizophrenia
There is little doubt that patients who are diagnosed with schizophrenia respond well to ECT. In addition, it is often assumed that most patients with schizophrenia who failed to respond to ECT are in fact misdiagnosed manics [47]. These doubts result from the difficulty in diagnosing schizophrenia. Nevertheless, most of the older studies of ECT on patients with schizophrenia are methodologically defective. [47]
Risks of electroconvulsive therapy
It is the effects of ECT that have been raising the concern of many people involved in the healthcare environment. Given the nature of the procedure, which involves applying electrical current to the patient’s head, one can see why such procedures might be considered dangerous. Yet studies have shown the opposite is true. In fact, according to the Royal Australian and New Zealand College of Psychiatrists (RANZCP), ECT is one of the safest and most effective treatments and is superior to drugs and placebos in the treatment of depressive illness [51].
Although ECT is safe to use and the mortality rate with the use of ECT is only two to four deaths per 100,000 treatments (considered a relatively low death rate) [51], other studies have shown different results. According to [52], two levels of side effects occur: short-term effects and long-term effects. Short-term effects include headache, confusion and memory disturbance. Long-term effects include permanent memory problems such as memory loss of past events and, with some people, memory loss of significant events. [46, 51, 52]
ECT and regulatory agencies
In the U.S., since the Medical Devices Act became law in 1976, many arguments have taken place between the ECT industry and ECT survivors. These are due to the fact that ECT devices never underwent any tests for safety under the FDA guidance. [53]
Even though ECT devices never underwent any tests for safety under the FDA guidance in the U.S., the FDA has approved ECT, though with limitations on energy levels and ranges. This has made it impossible to deliver the required level of energy for treating elderly patients who need to be treated with high energy, since the seizure threshold increases substantially with age. However, in Canada and Europe, ECT devices have been approved but with double the energy allowable by the U.S. devices. [46]
Currently, the FDA classifies ECT devices as class III even though they have not gone through the precise FDA testing that is usually required for medical devices, including the safety and efficacy tests. The only way that regulation is involved with ECT in the U.S. is through state governments, which vary widely. [49]
Public and professional doubts about ECT
Nowadays, ECT is viewed from one of two sides. The first side comprises ECT supporters, who support the use of ECT and mainly come from the environment of healthcare providers, including psychiatrists and, naturally, the people involved in the manufacture of ECT-related products. Their opponents include ECT survivors who do not support the use of ECT.
ECT is considered one of the most controversial treatments in medicine. It has an obvious history of misuse and abuse. Some of its survivors recall being given ECT without anaesthesia or muscle relaxants and live with nightmarish memories of the treatments. Others remember being punished with ECT for their perceived poor behaviour, or when forced to exhibit acceptable, normal behaviour. When these people describe their experiences with ECT today, they refer to the use of ECT as a torture. Moreover, some cultures have tended to represent the procedure of ECT in a negative light. [54]
Currently, many organizations exist to support ECT survivors such as the World Association of Electroshock Survivors, the Committee for Truth in Psychiatry and the larger Support Coalition International. Because the FDA had planned to reclassify ECT as a class II medical device, which would place ECT in a safe category, Dr. John Breeding, founder of the World Association of Electroshock Survivors, sent a letter to the FDA urging them not to change the classification. [55]
On the other hand, Professor Max Fink, a psychiatrist from the State University of New York, represented ECT supporters and did not hesitate to write a letter to the FDA supporting their action when he heard about the FDA reclassification of ECT. [56]
Regulatory Issues for Medical Devices that Apply Energy to Brain
In this chapter, there will be a discussion about issues on the requirements of regulatory agencies when dealing with the approval of different medical devices that apply energy to brain. There will be a measurement of those regulations’ effectiveness when dealing with the most important organ of the human body, that is, the brain. The reason for this discussion is to demonstrate whether the regulatory agencies should release such devices onto the market without any limitations or conditions for their use. This chapter highlights some of the issues in releasing the devices for public use and post market control of the performance of medical devices.
In this section, medical devices are categorized into two categories. The first comprises the normal-risk medical devices, which are devices mainly classified as class II medical devices. The discussion concerns ultrasound machines and their regulation. The reason for having chosen this device specifically is that the ultrasound’s effect is normal when it operates at the approved acoustic output, but when operating with a higher output, the ultrasound can cause serious harm to patients. In addition, ultrasound works with almost the same principles as TMS, which is safe to apply to a patient within the approved limit but, when the amount of energy delivered to the patient increases, can cause serious complications to patients. The second category includes the high-risk medical devices, which are the devices classified as class III medical devices. The discussion here will be concern TMS and DBS.
Regulations Governing Medical Devices
The safety and performance of medical devices depend largely on the critical elements of pre-market review which is concerned with the product control during design and development stage and post-market surveillance where the continued safety and effectiveness of the device is monitored. There is an additional and important element which is associated with the safety of the product is the representation of the product by the manufacturer to the user which is controlled by the labeling of the device. The objective of premarket control is to ensure that the device to be introduced in the market has complied with all regulatory requirements. Product representation is controlled by labeling and advertising control and this ensures that there is no misrepresentation on the use or functionality of the product to the ultimate user. Placing on-market control includes the processes of ensuring that the establishment is registered, the device is tested and confirming that it would meet all the after-sales obligations. Post-market surveillance is to ensure that the device meets the standards in respect of safety and performance continuously.
The following table provides a common framework for the regulations concerning the medical devices.
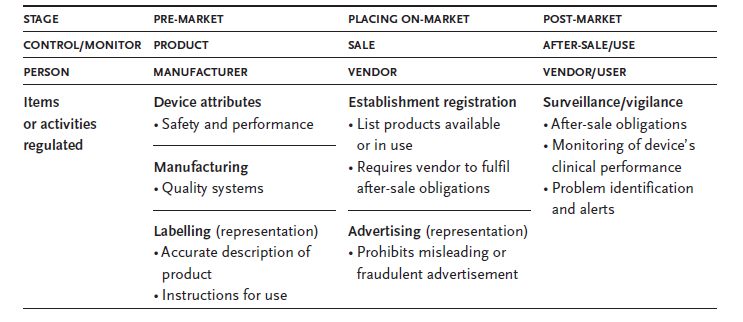
Different authorities in different countries of the world follow different systems and regimes for carrying out a pre-market review of the medical devices. However there is a common philosophy of meeting a satisfactory safety and performance standards is applied invariably by all the authorities. A minimum quality standard is also prescribed by them for marketing the devices. Specialized labeling requirements are also prescribed by the authorities. However the degree of control through the regulatory measures increases with the increase in the potential risk of the medical device being offered to the market.
Authorities follow different systems of product clearance for the devices to be marketed. In Australia TGA issues a Australian Register of Therapeutic Goods (ARTG) number for passing the devices to be marketed. Canada follows a different system of issuing a Device License as the authority to market the device. This license is issued by the regulatory authority – Therapeutic Products Directorate. In the EU the manufacturer has to obtain the EC certificate from a notified authority. On obtaining the certificate the manufacturer has to place the CE mark on the or within the device. In the United States the FDA issues a Marketing Clearance (510k) or an Approval Letter (PMA) to the manufacturer of the device enabling the marketing of the device.
In the United States most of the Class II and new devices which are different from the products eligible to be marketed legally and that does not require to apply for a Pre-Market Approval can obtain the clearance through PMA or Product Development Protocol processes. Most of class II and some of the Class I devices are cleared through the issue of a pre-market entry notification referred to as 510k. This form 510k is devised as an information package for the FDA and this form must demonstrate the ways in which the proposed medical device is substantially equivalent to any other medical device which is already existing in the US market. This route is less stringent than the PMA process. I am of the opinion that the approval through the submission of 510k form provides more room for unsafe devices entering the market and causing potential harm to the users. It would be of help to the patients, practitioners and users if the regulations provide for the display of the unpublished background information on the device as to the limitations in the use and the conditions subject to which the performance of the device can be expected as such information are vital to the users to safeguard from any potential hazard. However with the present regulatory requirement the manufacturers are under an obligation to provide only general information to the public knowledge and the clauses in form 510k requires the manufacturers to identify the ways in which the device proposed to be marketed is substantially equivalent to some other product being sold in the market. This information in my opinion is not sufficient enough to educate the users about the potential hazards that the device may cause. Therefore the display of unpublished information about the device should be made known to the prospective users to enable them fully understand the safety aspects of the device.
It is of critical importance that the safety and performance of the medical devices after they are marketed are continuously assessed. This becomes important in view of the fact that any amount of rigorous scrutiny in the pre-market trial would bring out all possible device failures or incidents caused by the misuse of the device. Only when the device is put to actual use the unforeseen problems with respect to the safety and performance of the devices would be thrown out. Therefore post-market surveillance comprising of “post-market surveillance studies” and “adverse event reporting” covering the monitoring of all the activities of medical devices in use assumes significance. In the post-market surveillance it is essential information is collected to ensure that the device satisfies the pre-approval conditions and to reaffirm that the use of the product does not pose any potential health hazard to the users.
The declarations of the manufacturers is the key to several medical devices as most of these fall in the category of medium to low risk classes with respect to their compliance with the regulations. Therefore there is the potential need for ensuring the quality standards of the device in all cases. This makes it vitally important for the manufacturer to conform to the quality standards and meet with all the regulatory requirements with respect to quality of the device being manufactured.
Public Release Issues
With the proliferation in the number of medical devices in the market it has become virtually impossible to ensure perfect performance of all the devices. Ensuring the continued safety of the medical devices has been one of the major challenges of FDA. Unsafe medical devices could easily cause trauma, injury and at times even may lead to fatal end of the patient using the device. Many of the common devices like defibrillators, pacemakers, infusion pumps, knee implants, hip replacements and tissue transplants may be found to have frequent issues in connection with the safety in using them. Such issues are commonly found even in some brand specific devices like Medtronic Defibrillators, Baxter Pumps and Guidant Defibrillators and pacemakers.
It is for the FDA to know at once about the failure of any device and to take prompt action to respond to the problems identified. This is essential to protect the consumers from the devices which do not function properly or are likely to result in potential harm to the users or patients causing injury or death. The Center for Devices and Radiological Health (CDRH) attached to FDA has assumed the responsibility for regulating medical devices by evaluating their safety and effectiveness before they are marketed and for monitoring their use throughout their life span after they are marketed. The amendments effected to Federal Food, Drug and Cosmetics Act in the year 1976 has prescribed a nationwide system called ‘post market surveillance’ to monitor the performance of marketed medical devices. However the medical devices which are in use to day are different in many respects from those used three centuries ago.
The challenges identified with the post marketing of medical devices arise from the nature of the technology used in the device, the increase in the number of devices being marketed and the difficulties surrounding the reporting of the problems associated with the adverse effects of devices. The major problem arises with the miniaturizing the devices so that more technology can be used. The manufacturers continue to modify the devices with a number of new features fitted into smaller spaces making the device smaller and this results in the increase in the number of parts which could potentially fail. Use of new technologies and stronger materials enlarge the useful life of the devices which also extends the chances of new problems to arise.
The proliferation of medical devices together with the complexity and the number of new types introduced in the market has impeded the efforts of FDA in monitoring the performance of medical devices. As of the year 2002 the medical device industry consisted of more than 15000 manufacturers producing and marketing about 100,000 individual medical devices covering various applications and uses. The increase in the volume of devices can be inferred from the fact that the number of pace makers has increased from 95,000 in the year 1990 to more than 267,000 in 2002. The sheer increase in volume poses a great challenge in terms of ensuring the safety of the devices. This challenge calls for stricter regulatory provisions to monitor the performance of the devices.
Normal-risk Medical Devices
The FDA has been regulating the ultrasound for a long time. The FDA believes that ultrasound requires much more detailed radiation data for 510(k) submissions. The FDA, through its Bureau of Radiological Health, has long regulated radiation-producing devices, such as X-rays, lasers, and ultrasound. Therapeutic ultrasound has had a regulatory standard for many years since it obviously causes biological effects. [57]. A specific regulation for therapeutic ultrasound arises from the contraindications that might result from ultrasound therapy, which have been listed in several publications [57, 58].
Excessive exposure can either lead to unnecessary risk or fail to achieve clinical benefit while inadequate exposure fails to achieve clinical benefit, resulting in the unnecessary exposure to ultrasound radiation. Since the intensity levels used in ultrasound therapy are in the range where adverse biological effects have been observed in animal studies, it is essential that the treatment doses are indicated and delivered accurately. To ensure that new devices can deliver prescribed exposures, the FDA regulates the maximum output of ultrasound devices to a prescribed level through a marketing approval process that requires devices be equivalent in efficacy and output to that acceptable range. [57]
This is exactly what one would expect not only of the FDA, but also of all other regulatory agencies. It is important to eliminate any error that may occur due to the inappropriate use of medical devices. In other words, a change in the regulation of acoustic output from medical ultrasound systems greatly increases the role the physician or sonographer plays in limiting the potential for ultrasound bioeffects. Because the maximum output limit was rather arbitrarily dictated by the FDA and because it might be diagnostically advantageous to increase this limit (i.e. since patients with large amounts of subcutaneous fat are difficult to scan), ultrasound devices are now being allowed to increase their output given sufficient feedback to the operator of the output level and its potential for biological effects. Therefore, the responsibility of an informed decision concerning the possible adverse effects of the ultrasound in comparison to the desired diagnostic information will likely become more important over the next few years. As it is currently envisioned, information would be provided to the operator concerning the relative potential for bioeffects and would allow the increase of acoustic output beyond a level that might induce a biological response.
High-risk Medical Devices
As mentioned earlier, high-risk medical devices are those classified as class III medical devices. The following discussion concerns the clearance of both TMS, which was recently cleared by the FDA [27], and DBS, which has been approved since 1997 [35]. Then, finally will conclude with a brief description of ECT.
Transcranial magnetic stimulation
The NeuroStar TMS Therapy system is the first and only TMS therapy device cleared by the FDA for the treatment of depression. To be approved, this therapy was evaluated for efficacy, safety and tolerability in the acute treatment of major depression in patients who had failed to receive benefit from prior antidepressant medications. This is exactly the purpose for which the FDA cleared the NeuroStar. [59]
The FDA supported its decision after evaluating the clinical trials that were provided by the device’s manufacturer. The issue that needs to be raised concerns the existence of other evidence that supports the FDA’s decision. According to [27], [59] and [18], not many differences exist between the clearance of this device and other class III devices, but there are differences in the results of the performed clinical trials. The other official requirements are exactly the same with all class III devices, which is exactly what needs to be reconsidered in order to achieve the maximum safety of patients.
Here, the regulatory agency should apply a certain condition in the approval. For example, the FDA, when it cleared the TMS system, should have considered the amount of energy that is to be delivered to the patient. This should eliminate the contraindications that might result from increasing the amount of magnetic field delivered to the patient. Furthermore, as seen in [60], [18], [30], [29] and [26], the maximum energy that should applied to the patient before he or she is hurt or that may lead to any damages is not mentioned.
However, the FDA clearance comes with the condition that TMS to be used with adult patients with major depression and failed to show improvement after receiving a single antidepressant. That was the only published condition that associates with TMS clearance. There are many issues that need to be considered with the clearance of TMS. For examble, from the side of health providing, who should use devices on patients? Is it the doctor or the technician? In addition, what would be the results if used on children or on elders?
Deep brain stimulation
There are always possible risks associated with the implantation of devices in humans, particularly when the implantation is in the brain. Thus, when dealing with DBS, the FDA considers it a class III medical device because its intended use as an implantable makes it a significant risk device.
DBS is increasingly becoming commercially utilized as a result of its emergence as an alternative to many therapeutic approaches and an adjunct to pharmacological involvements for some neurological disorders. However, the applications that have been approved for DBS are only for the treatment of Parkinson’s disease and dystonia; the rest of its applications are still in process. [42]
As previously mentiond, FDA has approved DBS to treat Parkinson’s and to treat dystonia but under the Humanitarian Device Exemption. According to [61], ” An Humanitarian Use Device is a device that is intended to benefit patients by treating or diagnosing a disease or condition that affects or is manifested in fewer than 4,000 individuals in the United States per year”. However, to obtain FDA approval for humanitarian use device, manufacturers should submit the Humanitarian Device Exemption application which is similar to the Premarket Approval application (PMA) but exempt from the effectiveness requirements[61]. That issue lead us to think about the cost of technology when dealing with patents and R&D. Because DBS is serving less than 4,000 individuals every year in US (which is very low percentage), FDA should be involved in pricing this technology especially when there is only one manufacturer for DBS.
Electroconvulsive therapy
According to a clinical trial that compared the efficacy of TMS and ECT in the treatment of severe depression [50], TMS is not as effective as ECT, and in the short-term treatment of depression, ECT is substantially more effective. Although it took a very short time for the FDA to clear TMS in comparison to ECT—which is approved but with the use of a very low level of energy—the reason for the difference in time to approval is not related to the efficacy of ECT but rather to the contraindications associated with the side effects of ECT.
Conclusion
It is the intention of the regulatory controls to safeguard the health and safety of all stakeholders including patients and users. The controls are extended by ensuring that the manufacturers of medical devices are obligated to follow certain specifided procedures during design, manufacturing and marketing of their devices.
It is vitally important that the regulatory controls are devised in proportion to the level of risks associated with the medical devices concerned. With the increase in the level of risks there should be corresponding increase in the level of regulatory controls. At the same time the control measures should take into account the benefits derived from the medical devices to the users and patients. It is also important that the regulatrory controls do not become a unnecessary burden either on the regulators or the manufacturers in the process of making the devices available for the practitioners’ use. It is to be understood that the risk associated with any particular device largely depends on the purpose intended to be met by the device. The effectiveness of risk management techniques applied during the design, manufacture and use also determine the degree of risk. The risk associated with the medical devices also depends to some extent on the intended users, the mode of its operation and the technologies involved. The regulatory controls should be able to take into account these factors especially they should provide for the technological advancements.
As seen in this minor thesis, there are many issues that need to be considered in relation to the approval of medical devices in general and those who apply a significant level of energy to the human brain in specific. These issues are varied between the premarket evaluation and the postmarketing surveillance.
As a young member of Saudi Food and Drugs Authority, I believe that regulatory agencies should reconsider the mechanisms of approving DBS, TMS, and ECT. In addition, due to the lack of information that related with the approval conditions which usually given by regulatory agencies to manufacturers, I suggest two things that need to be revised. First, FDA and other equivalent regulatory agencies should generously publish the full document of approval in order to allow public to be aware of all information that related with medical devices. Secondly, regulators should consider giving orders to manufacturers forcing them when publishing the approval or clearance they should show the conditions of approval and the limitation of use when existed. This will diminish any chance to misled public with such sensitive medical devices.
Without prejudice to any rules framed within their authority, the regulatory authorities would insist on the notification of the new devices being introduced in the market in their jurisdiction. The importance of such notification may be inferred from the use in assessing the evidence requirements for use in the conformity assessment procedures. Such notification may also help in reclassifying or to effect any change in the rules if there be any need for such revision or reclassification.
This thesis makes the following recommendations while applying regulatory controls to medical devices.
- Regulatory authorities may aim to fit the classification of the devices into a system that is globally recognized consisting of basic risk classes. This classification should allow for an efficient and graduated system of conformity assessment controls.
- The authorities may make the initial determination of class based on the features of the medical devices which create risk. The final classification in the case of most of the devices would normally follow the initial rules based classification.
- The regulatory controls should be capable of accommodating the future developments in the technology applied in the devices.
- The regulatory control authorities should insist on the manufacturer documenting the justification for placing the production of the devices into a particular risk class.
Alhough these recommendations are applied generally to all medical devices, it is important that all the factors affecting the risk classification in respect of the devices like the duration of device contact with the body and the degree of invasiveness involved need to be considered. There are other issues like whether the device is intended to deliver medicinal products or energy to the patient and whether the intended purpose of the device is to have a biological effect on the patient which affect the risk classification of the product. The device classification is also influenced by the local versus systemic effects of the device.
This thesis also reiterates that it is important to understand the purpose of risk classification in that such classification is essential to ensure that the regulatory controls are applied to a medical device in proportion to the risk it carries with respect to its use by the patient or any other person.
Medical Devices Market is huge and there are many experts who are offering their services to either manufacturers or regulators, but unfortunately, it is seldom to find one criticize the process that undergone to approve medical devices. This, I believe, is due to that experts are afraid of creating distances between them and regulators.
Finally, I challenge all medical devices experts who are involved in the regulatory process to be more generous in their publications in order to reach the best healthy environment. Note that, whenever the relation between manufacturers and regulators becoming more honest, this will reflect positively on patients and that was the main goal of the existence of both manufacturers and regulators.
List of References
“What Is GMP?,” GMP Institute. Web.
“Principles of Medical Devices Classification,” in Global Harmonization Task Force (GHTF): Study group No. 1, 2005.
“ISO Standards”. Web.
M. Cheng, “Medical Device Regulations : Global Overview and Guiding Principles,” Geneva : World Health Organization, 2003.
“ISO quality management standards.” Web.
“Medical Devices; Current Good Manufacturing Practice (CGMP) Final Rule; Quality System Regulation,” FDA, Ed. 1996.
G. Bartoo and G. Bartoo, “Regulatory issues [what is the QSR/GMP, and why should you care?] Regulatory issues [what is the QSR/GMP, and why should you care?],” Engineering in Medicine and Biology Magazine, IEEE, vol. 23, pp. 98-99, 2004.
R. Munzner, “Regulatory issues,” Engineering in Medicine and Biology Magazine, IEEE, vol. 23, pp. 207-208, 2004.
“Regulation of Medical Devices: Background Information for International Officials,” U.S. Department of Health and Human Services, FDA, 1999.
K. Sayre, J. Kenner, and P. L. Jones, “Safety models: an analytical tool for risk analysis of medical device systems,” in Computer-Based Medical Systems, 2001. CBMS 2001. Proceedings. 14th IEEE Symposium on, 2001, pp. 445-451.
G. Bartoo, “Risk management [medical devices],” Engineering in Medicine and Biology Magazine, IEEE, vol. 22, pp. 166-172, 2003.
ISO, “ISO 14971:2007 Document,” I. O. o. Sandards, Ed.: ISO, 2007.
R. P. Chiacchlerini and R. P. Chiacchlerini, “Medical Device Clinical Trial Design, Conduct, And Analysis Medical Device Clinical Trial Design, Conduct, And Analysis,” in Health Care Technology Policy I: The Role of Technology in the Cost of Health Care, 1994, 1994, pp. 278-285.
P. S. Saha, “Clinical trials of medical devices and implants: ethical concerns Clinical trials of medical devices and implants: ethical concerns,” Engineering in Medicine and Biology Magazine, IEEE, vol. 7, pp. 85-87, 1988.
Z. Wei Dong, Z. Wei Dong, and Y. Jing Hai, “The influence of transcranial magnetic stimulation on plasma β-endorphin in migraineurs The influence of transcranial magnetic stimulation on plasma β-endorphin in migraineurs,” in Engineering in Medicine and Biology Society, 2000. Proceedings of the 22nd Annual International Conference of the IEEE, 2000, pp. 1412-1413 vol.2.
T. Hortobagyi, T. Hortobagyi, and P. Bonato, “Transcranial magnetic stimulation Transcranial magnetic stimulation,” Engineering in Medicine and Biology Magazine, IEEE, vol. 24, pp. 20-21, 2005.
Neuronetics, “Neuronetics Transcranial Magnetic Stimulation,” Neuronetics, Ed. Web.
V. E. Amassian, V. E. Amassian, and P. J. Maccabee, “Transcranial Magnetic Stimulation Transcranial Magnetic Stimulation,” in Engineering in Medicine and Biology Society, 2006. EMBS ’06. 28th Annual International Conference of the IEEE, 2006, pp. 1620-1623.
J. Borckardt, A. Smith, S. Reeves, M. Weinstein, F. A. Kozel, Z. Nahas, N. Shelley, R. K. Branham, K. J. Thomas, and M. S. George, “Fifteen minutes of left prefrontal repetitive transcranial magnetic stimulation acutely increases thermal pain thresholds in healthy adults,” Pain research & management, vol. 12, pp. 287-90, 2007.
A. D. A. D. Wu, “Functional neuroimaging and repetitive transcranial magnetic stimulation in Parkinson’s disease,” Reviews in neurological diseases, vol. 4, pp. 1-9, 2007.
P. B. P. B. Fitzgerald and Z. J. Z. J. Daskalakis, “The use of repetitive transcranial magnetic stimulation and vagal nerve stimulation in the treatment of depression,” Current opinion in psychiatry, vol. 21, pp. 25-9, 2008.
B. B. Langguth, R. R. Wiegand, A. A. Kharraz, M. M. Landgrebe, J. J. Marienhagen, U. U. Frick, G. G. Hajak, and P. P. Eichhammer, “Pre-treatment anterior cingulate activity as a predictor of antidepressant response to repetitive transcranial magnetic stimulation (rTMS),” Neuro endocrinology letters, vol. 28, pp. 633-8, 2007.
W. W. H. Theodore and R. R. Fisher, “Brain stimulation for epilepsy,” Acta neurochirurgica. Supplementum, vol. 97, pp. 261-72, 2007.
T. Schlaepfer, “International Society for Transcranial Stimulation.” Web.
J. P. J. P. O’Reardon, H. B. H. B. Solvason, P. G. P. G. Janicak, S. S. Sampson, K. E. K. E. Isenberg, Z. Z. Nahas, W. M. W. M. McDonald, D. D. Avery, P. B. P. B. Fitzgerald, C. C. Loo, M. A. M. A. Demitrack, M. S. M. S. George, and H. A. H. A. Sackeim, “Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multisite randomized controlled trial,” Biological Psychiatry, vol. 62, pp. 1208-16, 2007.
Anonymous, “Clinical Neuroscience; FDA Clears Neurostar TMS Therapy for the Treatment of Depression,” Medical Letter on the CDC &FDA, Atlanta, 2008.
Brainsway, “Brainsway Transcranial Magnetic Stimluation,” B. Inc., Ed. Web.
“Positive Results Reported for Deep TMS H System For Depression,” Medgadget (Internet Journal of Emerging Medical Technologies), vol. Tuesday, 2008.
Neuronetics, “Durability of Acute Response to TMS in the Treatment of Major Depression: Relapse During a Continuation Pharmacotherapy Extension Study,” Neuronetics. Web.
M. Ogiue-Ikeda, M. Ogiue-Ikeda, S. Kawato, and S. Ueno, “The effect of transcranial magnetic stimulation on long-term potentiation in rat hippocampus The effect of transcranial magnetic stimulation on long-term potentiation in rat hippocampus,” Magnetics, IEEE Transactions on, vol. 39, pp. 3390-3392, 2003.
R. R. Bhidayasiri, T. T. Srikijvilaikul, C. C. Locharernkul, K. K. Phanthumchinda, and S. S. Kaoroptham, “State of the art: deep brain stimulation in Parkinson’s disease,” Medical journal of the Medical Association of Thailand, vol. 89, pp. 390-400, 2006.
S. SONG, “How Deep-Brain Stimulation Works,” Time vol. Sunday, 2006.
F. L. H. Gielen and F. L. H. Gielen, “Deep brain stimulation: current practice and challenges for the future Deep brain stimulation: current practice and challenges for the future,” in Neural Engineering, 2003. Conference Proceedings. First International IEEE EMBS Conference on, 2003, pp. 489-491.
Medtronic, “Deep Brain Stimulation,” in Activa. Web.
S. G. S. Holly A., Andrew G. S., “Reliability in Deep Brain Stimulation,” Device and material reliability, vol. 5, pp. 445 – 448, 2005.
S. K. Moore and S. K. Moore, “Psychiatry’s shocking new tools [brain stimulation techniques] Psychiatry’s shocking new tools [brain stimulation techniques],” Spectrum, IEEE, vol. 43, pp. 24-31, 2006.
MedicineNet.com, “Deep Brain Stimulation,” 2002.
M. S. Cameron C. McIntyre, Benjamin L. Walter, and Jerrold L. Vitek, “Mechanisms of Deep Brain Stimulation,” Journal of Clinical Neurophysiology, vol. 21, pp. 40-50, 2004.
S. Tisch, S. Tisch, J. C. Rothwell, P. Limousin, M. I. A. H. M. I. Hariz, and D. M. A. C. D. M. Corcos, “The Physiological Effects of Pallidal Deep Brain Stimulation in Dystonia.
The Physiological Effects of Pallidal Deep Brain Stimulation in Dystonia,” Neural Systems and Rehabilitation Engineering, IEEE Transactions on [see also IEEE Trans. on Rehabilitation Engineering], vol. 15, pp. 166-172, 2007.
C. Pea, K. Bowsher, A. Costello, R. A. De Luca, S. A. Doll, K. A. Li, M. A. Schroeder, and T. A. Stevens, “An Overview of FDA Medical Device Regulation as it Relates to Deep Brain Stimulation Devices,” Neural Systems and Rehabilitation Engineering, IEEE Transactions on [see also IEEE Trans. on Rehabilitation Engineering], vol. 15, pp. 421-424, 2007.
FDA, “Medtronic Activa® Dystonia Therapy – H020007,” C. f. D. a. R. Health, Ed.Web.
Medtronic, “Deep Brain Stimulation for the Treatment of Neurological Movement Disorders.” Web.
Medtronic, “Medtronic Activa Therapy Clinical Summary.” Web.
M. Fink and M. A. Taylor, “Electroconvulsive Therapy: Evidence and Challenges,” The Journal of American Medical Association, vol. 298, pp. 330-332, 2007.
R. Abrams and W. B. Essman, “Electroconvulsive Therapy: Biological Foundation and Clinical Applications,” Lancaster, England: MTP Press Limited, 1982.
R. Abrams, Electroconvulsive Therapy, 3 ed. vol. 1: Oxford University Press, Inc., New York, 1997.
T. C. Leinbaugh, “Electroconvulsive Therapy: A Primer for Mental Health Counselors,” Journal of Mental Health Counseling, vol. 2001, p. 2.
S. Eranti, A. Mogg, G. Pluck, and S. Landau, “A Randomized, Controlled Trial With 6-Month Follow-up of Repetitive Transcranial Magnetic Stimulation and Electroconvulsive Therapy for Severe Depression,” The American Journal of Psychiatry, vol. 164, pp. 73-81, Washington: 2007.
J. Little and G. Petrides, “Proceedings of the first Asia Pacific ECT Conference (Technical Aspects of ECT in Australia: Low, History and Practice),” Ballarat Health Services, 2001, p. 60.
C. H. Kellner, R. G. Knapp, G. Petrides, T. A. Rummans, M. M. Husain, and K. Rasmussen, “Continuation Electroconvulsive Therapy vs Pharmacotherapy for Relapse Prevention in Major Depression: A Multisite Study From the Consrtium for Research in Electroconvulsive Therapy (CORE),” The American Medical Association, vol. 63, pp. 1337-1344, 2006.
“The FDA and Electroconvulsive Therapy.” vol. 2008: ECT Organization, 2007.
V. Flint, “The Place of ECT in Mental Health Care,” Kai Tiaki: Nursing New Zealand, vol. 11, pp. 18-19, 2005.
J. Breeding, “Letter From World Association of Electroshock Survivors to FDA.” vol. 2008, 2004.
M. Fink, “Letter to FDA that Support the Reclassification of ECT.” vol. 2008 New York, 1990.
G. H. Myers, “FDA Regulation of Diagnostic Ultrasound,” in IEEE 1985 Ultrasonics Symposium, 1985, pp. 652-655.
J. F. Lehmann and B. J. d. Lateur, Therapeutic Heat and Cold vol. 1. Baltimore: Williams and Wilkins, 1982.
Anonymous, “FDA Clears Neurostar® TMS Therapy For The Treatment Of Depression,” in Medical News Today, 2008. Web.
Neuronetics, “Transcranial Magnetic Stimulation: Effectiveness and Safety in a Randomized, Controlled Multisite Trial and an Open-Label Extension Study,” Neuronetics. 2006. Web.
FDA, “Humanitarian Device Exemption,” 2008. Web.
Footnotes
“What Is GMP?,” GMP Institute. Web.
“Principles of Medical Devices Classification,” in Global Harmonization Task Force (GHTF): Study group No. 1, 2005.
“ISO Standards”. Web.
M. Cheng, “Medical Device Regulations : Global Overview and Guiding Principles,” Geneva : World Health Organization, 2003.
“ISO quality management standards”. Web.
“Medical Devices; Current Good Manufacturing Practice (CGMP) Final Rule; Quality System Regulation,” FDA, Ed. Web.
G. Bartoo and G. Bartoo, “Regulatory issues [what is the QSR/GMP, and why should you care?] Regulatory issues [what is the QSR/GMP, and why should you care?],” Engineering in Medicine and Biology Magazine, IEEE, vol. 23, pp. 98-99, 2004.
R. Munzner, “Regulatory issues,” Engineering in Medicine and Biology Magazine, IEEE, vol. 23, pp. 207-208, 2004.
“Regulation of Medical Devices: Background Information for International Officials,” U.S. Department of Health and Human Services, FDA, 1999.
K. Sayre, J. Kenner, and P. L. Jones, “Safety models: an analytical tool for risk analysis of medical device systems,” in Computer-Based Medical Systems, 2001. CBMS 2001. Proceedings. 14th IEEE Symposium on, 2001, pp. 445-451.
G. Bartoo, “Risk management [medical devices],” Engineering in Medicine and Biology Magazine, IEEE, vol. 22, pp. 166-172, 2003.
ISO, “ISO 14971:2007 Document,” I. O. o. Sandards, Ed.: ISO, 2007.
R. P. Chiacchlerini and R. P. Chiacchlerini, “Medical Device Clinical Trial Design, Conduct, And Analysis Medical Device Clinical Trial Design, Conduct, And Analysis,” in Health Care Technology Policy I: The Role of Technology in the Cost of Health Care, 1994, 1994, pp. 278-285.
P. S. Saha, “Clinical trials of medical devices and implants: ethical concerns Clinical trials of medical devices and implants: ethical concerns,” Engineering in Medicine and Biology Magazine, IEEE, vol. 7, pp. 85-87, 1988.
Z. Wei Dong, Z. Wei Dong, and Y. Jing Hai, “The influence of transcranial magnetic stimulation on plasma β-endorphin in migraineurs The influence of transcranial magnetic stimulation on plasma β-endorphin in migraineurs,” in Engineering in Medicine and Biology Society, 2000. Proceedings of the 22nd Annual International Conference of the IEEE, 2000, pp. 1412-1413 vol.2.
T. Hortobagyi, T. Hortobagyi, and P. Bonato, “Transcranial magnetic stimulation Transcranial magnetic stimulation,” Engineering in Medicine and Biology Magazine, IEEE, vol. 24, pp. 20-21, 2005.
Neuronetics, “Neuronetics Transcranial Magnetic Stimulation,” Neuronetics, Ed. Web.
V. E. Amassian, V. E. Amassian, and P. J. Maccabee, “Transcranial Magnetic Stimulation Transcranial Magnetic Stimulation,” in Engineering in Medicine and Biology Society, 2006. EMBS ’06. 28th Annual International Conference of the IEEE, 2006, pp. 1620-1623.
J. Borckardt, A. Smith, S. Reeves, M. Weinstein, F. A. Kozel, Z. Nahas, N. Shelley, R. K. Branham, K. J. Thomas, and M. S. George, “Fifteen minutes of left prefrontal repetitive transcranial magnetic stimulation acutely increases thermal pain thresholds in healthy adults,” Pain research & management, vol. 12, pp. 287-90, 2007.
A. D. A. D. Wu, “Functional neuroimaging and repetitive transcranial magnetic stimulation in Parkinson’s disease,” Reviews in neurological diseases, vol. 4, pp. 1-9, 2007.
P. B. P. B. Fitzgerald and Z. J. Z. J. Daskalakis, “The use of repetitive transcranial magnetic stimulation and vagal nerve stimulation in the treatment of depression,” Current opinion in psychiatry, vol. 21, pp. 25-9, 2008.
B. B. Langguth, R. R. Wiegand, A. A. Kharraz, M. M. Landgrebe, J. J. Marienhagen, U. U. Frick, G. G. Hajak, and P. P. Eichhammer, “Pre-treatment anterior cingulate activity as a predictor of antidepressant response to repetitive transcranial magnetic stimulation (rTMS),” Neuro endocrinology letters, vol. 28, pp. 633-8, 2007.
W. W. H. Theodore and R. R. Fisher, “Brain stimulation for epilepsy,” Acta neurochirurgica. Supplementum, vol. 97, pp. 261-72, 2007.
T. Schlaepfer, “International Society for Transcranial Stimulation”. Web.
J. P. J. P. O’Reardon, H. B. H. B. Solvason, P. G. P. G. Janicak, S. S. Sampson, K. E. K. E. Isenberg, Z. Z. Nahas, W. M. W. M. McDonald, D. D. Avery, P. B. P. B. Fitzgerald, C. C. Loo, M. A. M. A. Demitrack, M. S. M. S. George, and H. A. H. A. Sackeim, “Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multisite randomized controlled trial,” Biological Psychiatry, vol. 62, pp. 1208-16, 2007.
Anonymous, “Clinical Neuroscience; FDA Clears Neurostar TMS Therapy for the Treatment of Depression,” Medical Letter on the CDC &FDA, Atlanta Oct 26, 2008.
Brainsway, “Brainsway Transcranial Magnetic Stimluation,” B. Inc., Ed. Web.
“Positive Results Reported for Deep TMS H System For Depression,” Medgadget (Internet Journal of Emerging Medical Technologies), vol. Tuesday, 2008.
Neuronetics, “Durability of Acute Response to TMS in the Treatment of Major Depression: Relapse During a Continuation Pharmacotherapy Extension Study,” Neuronetics. Web.
M. Ogiue-Ikeda, M. Ogiue-Ikeda, S. Kawato, and S. Ueno, “The effect of transcranial magnetic stimulation on long-term potentiation in rat hippocampus The effect of transcranial magnetic stimulation on long-term potentiation in rat hippocampus,” Magnetics, IEEE Transactions on, vol. 39, pp. 3390-3392, 2003.
R. R. Bhidayasiri, T. T. Srikijvilaikul, C. C. Locharernkul, K. K. Phanthumchinda, and S. S. Kaoroptham, “State of the art: deep brain stimulation in Parkinson’s disease,” Medical journal of the Medical Association of Thailand, vol. 89, pp. 390-400, 2006.
S. SONG, “How Deep-Brain Stimulation Works,” Time vol. Sunday, 2006.
F. L. H. Gielen and F. L. H. Gielen, “Deep brain stimulation: current practice and challenges for the future Deep brain stimulation: current practice and challenges for the future,” in Neural Engineering, 2003. Conference Proceedings. First International IEEE EMBS Conference on, 2003, pp. 489-491.
Medtronic, “Deep Brain Stimulation”. Web.
S. G. S. Holly A., Andrew G. S., “Reliability in Deep Brain Stimulation,” Device and material reliability, vol. 5, pp. 445 – 448, 2005.
S. K. Moore and S. K. Moore, “Psychiatry’s shocking new tools [brain stimulation techniques] Psychiatry’s shocking new tools [brain stimulation techniques],” Spectrum, IEEE, vol. 43, pp. 24-31, 2006.
MedicineNet.com, “Deep Brain Stimulation”. Web.
M. S. Cameron C. McIntyre, Benjamin L. Walter, and Jerrold L. Vitek, “Mechanisms of Deep Brain Stimulation,” Journal of Clinical Neurophysiology, vol. 21, pp. 40-50, 1, 2004.
S. Tisch, S. Tisch, J. C. Rothwell, P. Limousin, M. I. A. H. M. I. Hariz, and D. M. A. C. D. M. Corcos, “The Physiological Effects of Pallidal Deep Brain Stimulation in Dystonia.
The Physiological Effects of Pallidal Deep Brain Stimulation in Dystonia,” Neural Systems and Rehabilitation Engineering, IEEE Transactions on [see also IEEE Trans. on Rehabilitation Engineering], vol. 15, pp. 166-172, 2007.
C. Pea, K. Bowsher, A. Costello, R. A. De Luca, S. A. Doll, K. A. Li, M. A. Schroeder, and T. A. Stevens, “An Overview of FDA Medical Device Regulation as it Relates to Deep Brain Stimulation Devices,” Neural Systems and Rehabilitation Engineering, IEEE Transactions on [see also IEEE Trans. on Rehabilitation Engineering], vol. 15, pp. 421-424, 2007.
FDA, “Medtronic Activa® Dystonia Therapy – H020007,” C. f. D. a. R. Health, Ed. Web.
Medtronic, “Deep Brain Stimulation for the Treatment of Neurological Movement Disorders”. Web.
Medtronic, “Medtronic Activa Therapy Clinical Summary”. Web.
M. Fink and M. A. Taylor, “Electroconvulsive Therapy: Evidence and Challenges,” The Journal of American Medical Association, vol. 298, pp. 330-332, 2007.
R. Abrams and W. B. Essman, “Electroconvulsive Therapy: Biological Foundation and Clinical Applications,” Lancaster, England: MTP Press Limited, 1982.
R. Abrams, Electroconvulsive Therapy, 3 ed. vol. 1: Oxford University Press, Inc., New York, 1997.
T. C. Leinbaugh, “Electroconvulsive Therapy: A Primer for Mental Health Counselors,” Journal of Mental Health Counseling, vol. 2001, p. 2.
S. Eranti, A. Mogg, G. Pluck, and S. Landau, “A Randomized, Controlled Trial With 6-Month Follow-up of Repetitive Transcranial Magnetic Stimulation and Electroconvulsive Therapy for Severe Depression,” The American Journal of Psychiatry, vol. 164, pp. 73-81, Washington: 2007.
J. Little and G. Petrides, “Proceedings of the first Asia Pacific ECT Conference (Technical Aspects of ECT in Australia: Low, History and Practice),” Ballarat Health Services, 2001, p. 60.
C. H. Kellner, R. G. Knapp, G. Petrides, T. A. Rummans, M. M. Husain, and K. Rasmussen, “Continuation Electroconvulsive Therapy vs Pharmacotherapy for Relapse Prevention in Major Depression: A Multisite Study From the Consrtium for Research in Electroconvulsive Therapy (CORE),” The American Medical Association, vol. 63, pp. 1337-1344, 2006.
“The FDA and Electroconvulsive Therapy.” vol. 2008: ECT Organization, 2007.
V. Flint, “The Place of ECT in Mental Health Care,” Kai Tiaki: Nursing New Zealand, vol. 11, pp. 18-19, 2005.
J. Breeding, “Letter From World Association of Electroshock Survivors to FDA.” vol. 2008, 2004.
M. Fink, “Letter to FDA that Support the Reclassification of ECT.” vol. 2008 New York, 1990.
G. H. Myers, “FDA Regulation of Diagnostic Ultrasound,” in IEEE 1985 Ultrasonics Symposium, 1985, pp. 652-655.
J. F. Lehmann and B. J. d. Lateur, Therapeutic Heat and Cold vol. 1. Baltimore: Williams and Wilkins, 1982.
Anonymous, “FDA Clears Neurostar® TMS Therapy For The Treatment Of Depression,” in Medical News Today. Web.
Neuronetics, “Transcranial Magnetic Stimulation: Effectiveness and Safety in a Randomized, Controlled Multisite Trial and an Open-Label Extension Study,” Neuronetics. Web.
FDA, “Humanitarian Device Exemption,” 2008. Web.